
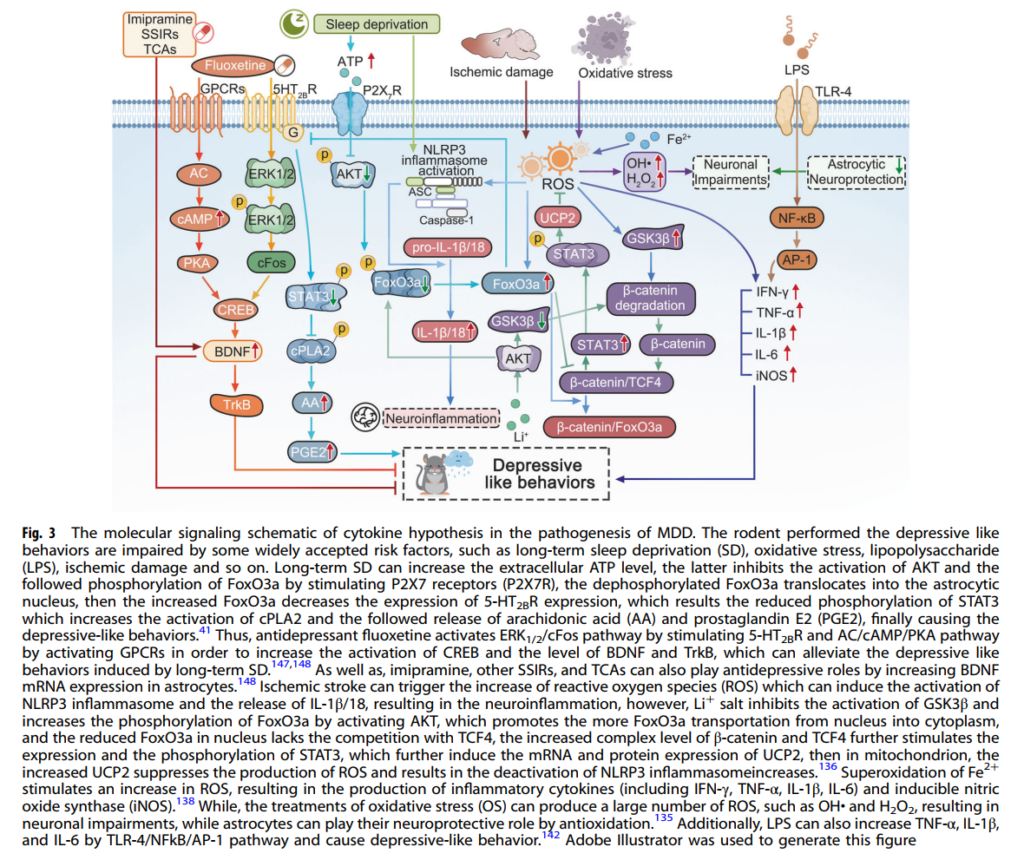
Fig. 3 MDD(大うつ病性障害)の発症におけるサイトカイン仮説の分子シグナル伝達模式図
齧歯類において、うつ病様行動を示す被験体は、長期の睡眠剥奪(SD)、酸化ストレス、リポ多糖(LPS)、虚血性損傷などの広く認められているリスク因子によって障害されます。
長期の睡眠剥奪は、細胞外ATPレベルを上昇させ、これがP2X7受容体(P2X7R)を刺激することでAKTの活性化およびその後のFoxO3aのリン酸化を抑制します。リン酸化が解除されたFoxO3aはアストロサイトの核内へ移行し、その結果、FoxO3aの増加により5-HT2B受容体の発現が低下します。これにより、STAT3のリン酸化が減少し、cPLA2の活性化およびその後のアラキドン酸(AA)とプロスタグランジンE2(PGE2)の放出が促進され、最終的にうつ病様行動が引き起こされます。41
これに対し、抗うつ薬であるフルオキセチンは、5-HT2B受容体を刺激してERK1/2/cFos経路を活性化し、またGPCRの活性化によってAC/cAMP/PKA経路を活性化することで、CREBの活性化およびBDNFとTrkBのレベルを上昇させ、長期SDによって誘発されるうつ病様行動を改善します。147,148
同様に、イミプラミン、その他のSSIR、およびTCAも、アストロサイトにおけるBDNF mRNAの発現を増加させることにより、抗うつ作用を発揮することが報告されています。148
虚血性脳卒中は、反応性酸素種(ROS)の増加を引き起こし、これがNLRP3インフラマソームの活性化およびIL-1β/18の放出を誘導して神経炎症を引き起こしますが、Li⁺塩はAKTを活性化することによりGSK3βの活性化を抑制し、FoxO3aのリン酸化を増加させます。これにより、FoxO3aが核から細胞質へさらに輸送され、核内のFoxO3aがTCF4との競合を欠くようになると、β-cateninとTCF4の複合体レベルの上昇がSTAT3の発現およびリン酸化をさらに刺激し、これがUCP2のmRNAおよびタンパク質発現を誘導します。すると、ミトコンドリア内でのUCP2の増加がROSの産生を抑制し、結果としてNLRP3インフラマソームの活性化が低下します。136
Fe²⁺の超酸化がROSの増加を促し、これにより炎症性サイトカイン(IFN-γ、TNF-α、IL-1β、IL-6など)および誘導型一酸化窒素合成酵素(iNOS)の産生が引き起こされます。138
一方、酸化ストレス(OS)の処理によって大量のROS、例えばOH・やH₂O₂が産生され、これが神経細胞の障害を引き起こしますが、アストロサイトは抗酸化作用を通じて神経保護を果たすことができます。135
さらに、LPSはTLR-4/NFkB/AP-1経路を介してTNF-α、IL-1β、およびIL-6の産生を増加させ、うつ病様行動を引き起こします。142
