
遺伝子制御ネットワークと転写制御に関する研究文書
最近の研究では、ヒトと霊長類に特異的な遺伝子制御ネットワークの側面も特定されており、これらはヒトと霊長類の皮質の独特な特性、そして高度なヒトの認知機能にとって不可欠である可能性が高い。例えば、シナプス形成因子であるCBLN2は、発達中の前頭前皮質において種特異的な発現差を示す。ヒトの中期胎児前頭前皮質では、CBLN2は皮質板(CP)の全層で発現するが、マウス前頭前皮質では、ヒトの中期胎児発達に相当する発達段階において、Cbln2の発現は上層に制限される。この発現の違いは、CBLN2の前頭前皮質エンハンサーにおけるヒト族(ヒト、チンパンジー、ボノボ)特異的な欠失によるものである(図1.3-4C)。これらの欠失領域には抑制性転写因子SOX5の結合部位が含まれており、ヒトの中期胎児前頭前皮質におけるCBLN2発現の脱抑制につながっている。マウスのCBLN2エンハンサーのin situ遺伝的ヒト化により、CBLN2のヒト様発現とPFC錐体ニューロンの樹状突起スパインの増加がもたらされた。注目すべきことに、ヒトのPFCの錐体ニューロンには、他の分析された霊長類のPFCと比較して不釣り合いに多くのスパインが存在する。
ヒト神経発達の転写制御
モデル生物における個々のCRE(シス制御エレメント)の研究は神経発達の現在の理解に大きな進歩をもたらしたが、ヒトの神経発達における遺伝子制御の包括的理解には、ヒトで活性を持つすべてのCREのカタログが必要であることも明らかになっている。進化過程でのゲノム配列の保存性に基づいて、多くの推定CREが特定されており、その大部分はヒト脳に豊富に存在する遺伝子の発現を制御すると提案されている。逆に、他の生物と比較してヒト系統で置換率が劇的に増加したDNA配列も、ヒトのCREを予測するために使用されている。

図1.3-5. A. ヒト進化エレメントの概略図。ヒト加速領域(HAR)は、他の霊長類と比較してヒトで配列変化が加速した領域として定義される。ヒト高メチル化領域は、他の霊長類と比較してヒトでDNAメチル化が増加した領域として定義される。ヒト獲得エレメント(HGE)は、他の霊長類と比較してヒトでH3K27acレベルによって定義される制御活性を獲得したゲノム領域として定義される。ヒト失失エレメント(HLE)は、ヒトで失われた制御領域として定義される。B. ヒト進化エレメントに関連する遺伝子は、胎児脳(FB)の外側放射状グリア細胞と内皮細胞、および成人脳(AB)のアストロサイトに豊富に存在する。(Won H, Huang J, Opland CK, et al. Human evolved regulatory elements modulate genes involved in cortical expansion and neurodevelopmental disease susceptibility. Nat Commun 2019;10(1):2396. doi: 10.1038/s41467-019-10248-3. License: https://creativecommons.org/licenses/by/4.0/ より転載)
転写物解析の研究と同様に、高スループット技術の開発(「分子ランドスケープを研究する高スループット技術」の項を参照)により、ヒト脳におけるCREの検出能力が大幅に向上した。従来、クロマチンアクセシビリティとヌクレオソーム欠失、そしてより最近では特定の翻訳後ヒストン修飾の濃縮が、CREの定義的特徴の一つと考えられてきた。トランスポザーゼアクセシブルクロマチン高スループットアッセイ(ATAC)とディープシーケンシングに続くChIPアッセイ(ChIP-seq)が死後ヒト脳組織に適用されている。Viselらは、ヒト胎児新皮質における活性エンハンサーを特定するため、エンハンサー関連タンパク質p300/CBPのゲノム全体での占有を決定した。ヒストンの2つのエンハンサー豊富な翻訳後修飾、ヒストン3リジン4モノメチル化(H3K4me1)とヒストン3リジン27アセチル化(H3K27ac)の濃縮をマッピングすることにより、異なる脳領域と異なる時点で活性なエンハンサーが解読された。例えば、Wonらは、ヒト加速領域、ヒト獲得エンハンサー、ヒト失失エンハンサーを含む、ヒトにおける進化的に動的な領域を特定した(図1.3-5A)。彼らはこれらの領域を潜在的標的遺伝子に注釈し、ヒト進化領域が成人皮質オリゴデンドロサイトと胎児外側放射状グリア細胞で発現する遺伝子に豊富であることを発見した(図1.3-5B)。ヒト皮質領域特異的ATAC-seq研究と組み合わせることで、これらの研究はヒトで獲得されたエンハンサーと皮質領域に特異的な推定エンハンサーを特定した。さらに、細胞型および文脈特異的ネットワークを形成する共制御エンハンサーが特定されている。これらのエンハンサーネットワークの多くは、神経細胞の増殖、移動、皮質マップ形成に機能する共発現遺伝子のモジュールと相関している。したがって、発達中のヒト中枢神経系における時空間的遺伝子発現を制御する制御ネットワークの解明が始まっているが、この情報はまだ完全に機能的文脈に位置づけられていない。
- CBLN2遺伝子の種特異的発現:ヒト族(ヒト、チンパンジー、ボノボ)に特異的な遺伝的変化により、前頭前皮質でのCBLN2の発現パターンがマウスと異なることを説明
- ヒト進化エレメント:ヒト加速領域(HAR)、ヒト獲得エレメント(HGE)、ヒト失失エレメント(HLE)などの概念を紹介
- 高スループット技術の応用:ATAC-seqやChIP-seqなどの技術を用いたヒト脳における遺伝子制御エレメントの同定方法
この研究は、ヒトの高次認知機能の進化的基盤を理解する上で重要な知見。
ヒト脳発達のエピジェネティック制御
エピジェネティクスとは、DNA配列を変化させることなく遺伝子発現を調節することができるDNA、クロマチン構造、RNAの化学的修飾を指す。エピジェネティック状態は神経発達において動的であり、ゲノムと環境の両方に影響される。したがって、エピジェネティック制御は精神疾患に対する感受性が生じるメカニズムである。最も一般的に研究されている2つのエピジェネティックメカニズムは、転写因子のDNA結合を阻害することにより一般的に遺伝子発現を抑制するDNAメチル化と、転写機構へのアクセシビリティを増減させるためにクロマチン構造を変化させる翻訳後ヒストン修飾である。
遺伝子プロモーターに豊富に存在する主にCpGヌクレオチドでのシトシンのDNAメチル化は、神経発達と機能において重要な役割を果たし、ヒトゲノムにおいて数十万から数百万のCpGを偏りなく解析することができる。胎児から若年成人の発達期間中の広範囲にわたるメチローム再構成が複数の研究で観察されている。ヒト前頭前皮質における全体的DNAメチル化は、胎児期から出生後の発達期間中に大幅に再構成されることが示された。これらの変化は胎児期の発達中により急速であり、出生後と加齢とともに減速した。これらのメチル化変化の多くは、発達期間中のヒト皮質の細胞構成変化と有意に相関し、ヒト神経発達におけるDNAメチル化の重要な役割を強調している。
CpGのメチル化に加えて、シナプス形成の主要段階と一致する出生後早期発達期間中の非CpGメチル化(mCH)の蓄積が最近ヒト脳で発見された。この蓄積は細胞型特異性を示し、ニューロンに豊富なmCHと、グリア細胞では無視できるレベルを示す。CHメチル化はヒト脳進化の過程で増加し、遺伝子と共発現モジュールのヒト特異的ダウンレギュレーションに寄与している。CH高メチル化の効果は特にニューロンサブタイプで顕著であり、ヒトの出生後早期発達で活性である;したがって、CHメチル化によって媒介される発達過程の破綻は神経精神疾患への脆弱性に寄与する可能性があり、それらを理解することはこれらの疾患に対する標的治療の導出に新たな洞察をもたらす可能性がある。CpGとmCHメチル化の両方について、隣接遺伝子の発現との強い相関が観察されている。
細胞多様性を確立し神経発達のタイミングを制御するもう一つの重要なエピジェネティックメカニズムは、ヒストンの翻訳後修飾である。メチル化(モノ、ジ、トリ)とアセチル化が最も広く研究されている修飾であるが、さらに多くの修飾が観察されている。研究では、次世代シーケンシングと結合したChIP(ChIP-seq)を適用することにより、限られた数の死後ヒト脳においてクロマチン状態マップが生成されている。Cheungらは、出生後0.5年から69年までの年齢範囲の11個体のヒト前頭前皮質のニューロンおよび非ニューロン細胞において、発達を通じてヒストン3リジン4トリメチル化(H3K4me3)をプロファイリングした。彼らの研究の重要な発見は、前頭大脳皮質ニューロンの出生後発達と加齢期間中の有意なリモデリングであった。DNAのメチル化と脱メチル化の両方がすべての哺乳類種においてニューロンの分化と機能に必要である;細胞がもはや分裂しなくなった出生後に、高レベルの5-ヒドロキシメチルシトシン(5hmC)が哺乳類ニューロンに蓄積することが観察されている。終末分化におけるまたは活性DNA脱メチル化の中間体としての5hmCの役割に関する我々の知識はまだ発展中である;最近、Stoyanovaらは動物モデルシステムにおいて、生後第1週以降に5hmCが継続的に産生されなければニューロンが適切に成熟しないことを実証した。彼らのデータは、5mCを5hmCに変換するTetタンパク質の役割を強調し、メチル化マークの除去に向けた準備と、これらのマークの活性除去がニューロン分化に必須であることを実証した。まとめると、これらのデータは、エピジェネティックメカニズムが遺伝子発現の地域的および時間的ランドスケープの確立において重要な役割を果たし、ヒト脳進化に寄与することを確認している。
神経発達障害および精神疾患における転写およびエピジェネティック制御異常
多くの神経精神疾患が早期神経発達の遺伝的および環境的摂動に根ざしていることがますます理解されるようになっている。過去数年間、遺伝学研究は精神疾患の遺伝的危険因子の特定において重要な進歩を遂げた。ほとんどの精神疾患は非常に複雑であり、複数の遺伝子が疾患リスクに寄与している。精神疾患に関与する遺伝的因子が一般的および稀な変異(SNP)と染色体構造の亜顕微鏡的変異(すなわちCNV)の両方を含むという非常に強い証拠がある。例えば、統合失調症と双極性障害では、遺伝子発見はこれらの疾患と一般的SNPとの連鎖に焦点を当てているが、自閉症スペクトラム障害では、高い浸透率を持つ稀でde novo単一ヌクレオチド変異が重篤な症候群を引き起こすことができ、通常知的障害を伴う。神経精神疾患が単一の原因遺伝子によって現れる場合、多くの神経発達障害(例:脆弱X症候群[FXS])のように、変異遺伝子の直接的生物学的役割とは独立して最終的に疾患表現型につながる神経発達過程の広範な制御異常がある。一般的精神疾患の多遺伝子性は、精神疾患の病態生理学の基礎となる分子メカニズムの特定を困難にしている。疾患の遺伝的基盤を理解する能力をさらに複雑にしているのは、上記の生殖細胞系変異に加えて、受精後に生じ、しばしば特定の細胞および組織型で起こるde novo変異である体細胞変異が精神および神経発達障害に寄与する可能性があることである。
遺伝学と複雑な多遺伝子リスク構造は、多くの精神疾患への感受性を完全に説明しない。実際、多くの疾患について、一卵性双生児の両方が疾患を発症する可能性は遺伝的リスクよりも低く、環境因子が疾患感受性に影響することを示している。例えば、産科合併症などの環境因子が統合失調症発症リスクを増加させることが報告されている。環境からの有害刺激は、遺伝子発現レベルの変化から神経活動の制御異常まで、発達中の脳の細胞過程に広範な影響を与え、これが神経発達と相互作用して精神的問題への感受性増加をもたらす。さらに、発達期の有害事象は遺伝子発現に永続的影響を与える可能性があるエピジェネティック変化につながる可能性がある;神経精神疾患状態では、確率的影響-典型的発達の経過に最小限しか影響しないランダムな事象-が重症度と転帰の変動に増幅された効果を持つ可能性がある。これは、高度に予測可能な応答ではなく高度に変動的な応答を誘導する方法で環境の変動に対する感受性を増加させる遺伝的変異の特定の例である。
この病因の複雑さにもかかわらず、精神疾患の基礎となる神経発達の破綻は、明確でおそらく定義可能な細胞および分子過程、脳回路、および発達の時代に収束する。これらの過程を解決することは、基礎となる病理のより良い理解をもたらし、より標的化された治療戦略につながる可能性がある。以下のいくつかの例で説明されるように、神経典型的ヒト神経発達の細胞および分子生物学のますます包括的な研究は、精神疾患、遺伝学、および病理学の研究と統合されて、疾患メカニズムを調査し、破綻した分子過程が作用する可能性の高い時期と場所を特定することができる。
脆弱X症候群:単一遺伝子変異が広範囲な下流結果をもたらす可能性
FXSは知的障害の主要な遺伝的原因であり、しばしば自閉症様特徴、攻撃性、注意欠陥、言語・発話発達の遅れを伴う。FXSは、mRNAの局在、安定性、翻訳に関与するRNA結合タンパク質(FMRP)をコードするFMR1遺伝子の機能喪失によって引き起こされる。したがって、FMRP機能の喪失は遺伝子発現と翻訳に広範囲な下流結果をもたらす。例えば、最近の研究では、FMRPが保存された過程と種依存的過程の両方を通じてヒト神経性一酸化窒素合成酵素1(NOS1)遺伝子と相互作用することが示された。大脳皮質では、NOS1タンパク質発現は第5層(L5)皮質下投射錐体ニューロンに限定され、前頭頭頂弁蓋部の交互ミニコラムでの発現を示す。ヒトでは、FMRPはNOS1コード領域の配列と相互作用し、NOS1タンパク質発現の増加につながる。一致して、新皮質NOS1タンパク質レベルは発達中のFXSヒト新皮質で劇的に減少しており、ここでNOS1とFMRPは言語、発話、複雑な社会行動に関与する領域の錐体ニューロンの亜集団においてシナプス形成期に一過性に共発現する。
ダウン症候群と自閉症スペクトラム障害:特定の発達過程に収束する多遺伝子破綻
ダウン症候群(DS)は、多遺伝子破綻の源(21番染色体トリソミー)が十分に確立されているため、多遺伝子性複雑疾患の生物学的結果を研究するための興味深い神経発達障害を提供する。研究では、21番染色体トリソミーが遺伝子発現のゲノム全体破綻を引き起こすことが示されている。これらの中で、DS発達脳転写物の特性化では、すべての染色体上の遺伝子の約5%が発達のある時点で制御異常を示し、21番染色体遺伝子の18%を除いて、ほぼ同数のアップレギュレートとダウンレギュレート遺伝子があり、21番染色体遺伝子はすべてアップレギュレートされていた(図1.3-6)。遺伝子共発現解析では、これまでDSに関連していた過程(例:シナプス機能とニューロン特定)に関与する遺伝子ネットワークと、ミエリン化とオリゴデンドロサイト発達に関連する遺伝子の発現変化など、DSでよく確立されていなかった特定の細胞および分子過程の遺伝子の異常が明らかになった。その後のDS死後ヒト脳標本の解析では、顕著な髄鞘形成不全、有髄線維の無秩序、有髄線維路を通じた伝導の遅延がDS神経病理学的特徴として明らかになった。
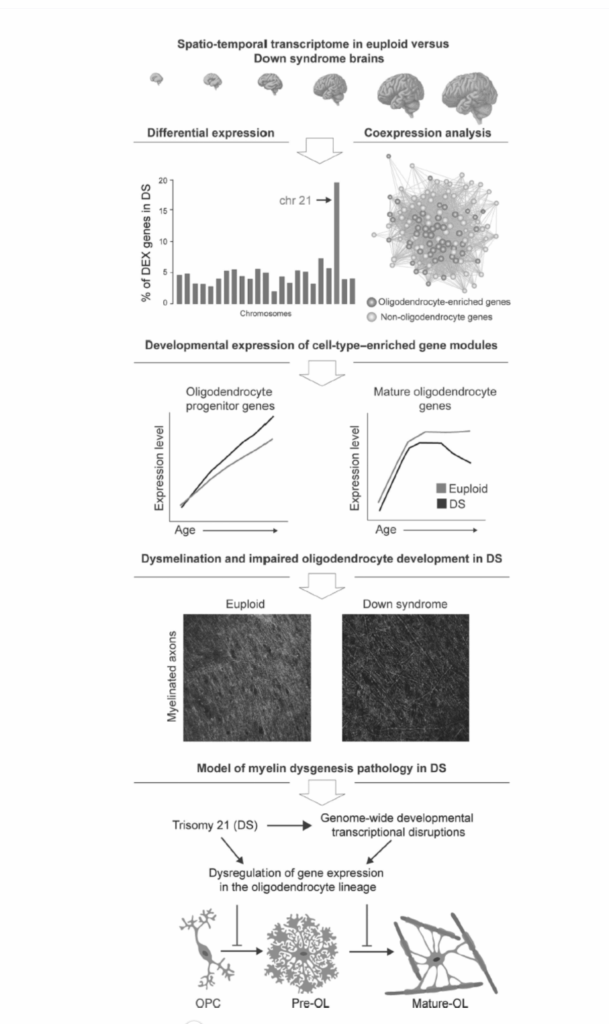
図1.3-6. ダウン症候群(DS)におけるゲノム全体制御異常がオリゴデンドロサイト発達障害を引き起こす。Olmos-Serranoらによる研究の概略図で、DS(21番染色体トリソミー、DS)の発達転写物特性化が多遺伝子性遺伝子破綻が遺伝子発現、最終的には細胞および分子機能により広範囲な影響をもたらす方法についての洞察を提供したことを示している。この研究では、中期胎児発達から成人期までのDS個体の複数の脳領域で全体的遺伝子発現が特性化された(第1パネル)。21番染色体遺伝子の約20%が発達のある時点で誤発現し、他のすべての染色体の遺伝子の3%から8%が誤発現することが決定された。したがって、総合的には、DSではるかに多くの非21番染色体遺伝子が誤発現している(第2パネル)。共発現遺伝子のネットワーク解析により、ミエリン化遺伝子に高度に濃縮されたモジュールが明らかになった。オリゴデンドロサイト前駆細胞または成熟オリゴデンドロサイトのいずれかで高発現する遺伝子に濃縮された遺伝子ネットワークのその後の解析により、オリゴデンドロサイト系統における発達遺伝子発現動態の実質的制御異常が明らかになった:前駆体遺伝子はより高発現し、成熟遺伝子はより低レベルで発現した(第3パネル)。前頭前皮質の有髄線維路の解析により、DSにおける髄鞘形成不全と有髄軸索の無秩序が明らかになった(第4パネル)。この研究は、21番染色体トリソミーが発達遺伝子制御のゲノム全体破綻を引き起こし、これは特にオリゴデンドロサイトで顕著であり、オリゴデンドロサイト成熟障害と髄鞘形成不全をもたらしたことを示唆している(第5パネル)。(Olmos-Serrano JL, Kang HJ, Tyler WA, et al. Down syndrome developmental brain transcriptome reveals defective oligodendrocyte differentiation and myelination. Neuron 2016;89(6):1208-1222. Copyright @ 2016, Elsevierからの許可を得て改変。doi: 10.1016/j.neuron.2016.01.042.)
同様に、ASDと統合失調症の病因を調査するため、最近の研究では計算アプローチを用いてGWASと全エクソームシーケンシングからのリスク変異データを時空間脳転写物データと統合している。例えば、これまでにASDと関連した遺伝子の共発現ネットワークを構築することにより、研究者は高度に関連した遺伝子のモジュールを特定し、これらの遺伝子ネットワークが濃縮される発達時期と脳の細胞型を決定した。興味深いことに、ASDリスク遺伝子は中期胎児発達期の前頭葉皮質投射ニューロンに高度に濃縮されることが発見された(図1.3-7)。注目すべきことに、統合失調症に関連する遺伝子も同様の濃縮を示した。これらのデータは、ASDと統合失調症において、皮質投射ニューロンが最終移動過程にあり樹状突起と初期軸索投射の精巧化を開始するときに発達が特に破綻する可能性があることを示している。さらに、機能的および解剖学的画像研究により、これらのニューロンで構成される脳回路がASDと統合失調症の両方で異常であることが明らかになった。
神経発達障害における非コード変異
疾患関連配列変異の大部分はゲノムの非コード領域に生じる。これらの変異の結果は研究が困難であるが、多くは発達効果を持ち、精神疾患のリスク因子を表す可能性が高い。これは特に、エンハンサーやプロモーターなどの制御領域内にある変異の場合である。説明的な例は、幹細胞増殖と大脳皮質発達にとって重要なGカップリングタンパク質受容体コード遺伝子であるGPR56の17個の転写開始部位(TSS)の1つの直上流の欠失から生じるシルビウス周囲多小脳回症の最近記述された研究である。5人の罹患個体で、出生前発達期間中の脳のシルビウス周囲領域での発現駆動に必須であるGPR56コアプロモーター領域で15塩基対欠失が検出された。この欠失は、特に神経形成期のシルビウス周囲皮質でのGPR56発現を障害し、前駆細胞増殖の減少と、特にシルビウス周囲皮質での小脳回形成につながった可能性が高い(図1.3-4B)。この例は極端であるが、非コード配列、特に制御エレメントの違いが異なる疾患感受性と神経発達表現型をもたらす方法を実証している。
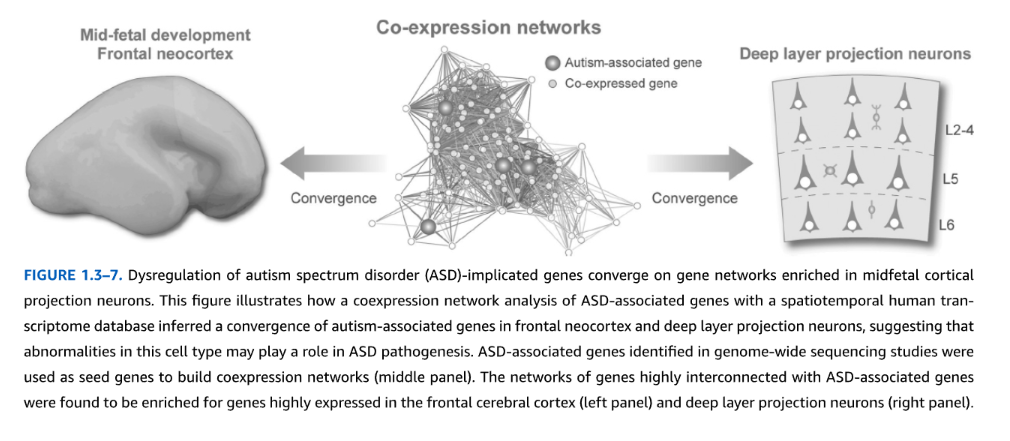
図1.3-7. 自閉症スペクトラム障害(ASD)関連遺伝子の調節異常は、中期胎児期皮質投射ニューロンで濃縮された遺伝子ネットワークに収束する。この図は、ASD関連遺伝子と時空間的ヒト転写産物データベースの共発現ネットワーク解析により、自閉症関連遺伝子が前頭新皮質と深層投射ニューロンに収束することが推定され、この細胞型の異常がASD病因に役割を果たしている可能性を示唆していることを表している。ゲノムワイド配列決定研究で特定されたASD関連遺伝子をシード遺伝子として用いて共発現ネットワークを構築した(中央パネル)。ASD関連遺伝子と高度に相互接続されている遺伝子のネットワークは、前頭大脳皮質(左パネル)と深層投射ニューロン(右パネル)で高発現する遺伝子に濃縮されていることが判明した。
体細胞変異:局所的に離散的な遺伝子変異と遺伝子誤発現のパターン
標準的なゲノムスクリーニング技術では脳内の体細胞変異を容易に検出することはできないが、深層次世代シーケンシングや単一細胞シーケンシングの最近の進歩により、特定の細胞や組織における体細胞変異の検出が可能となり、疾患病因学への理解が深まっている。この研究は、新しい疾患機序の解明から神経型発達への新たな洞察の提供まで、幅広い意味を持つ。例えば、これらの研究では、特定の神経発達疾患は神経前駆細胞におけるde novo体細胞変異によって引き起こされる可能性があることが実証されており、これらの変異はクローン性に拡大し、特定の神経細胞型や脳領域に異常を生じ、脳奇形やより微細な表現型を引き起こす可能性がある。これは統合失調症の死後研究によって最近示唆されている。
統合失調症におけるエピジェネティック調節異常
エピジェネティック機序の調節異常は、疾患状態で観察される転写産物の変化を引き起こす可能性がある。精神疾患におけるエピジェネティック修飾の役割の理解はまだ初期段階にあるが、最近の系統的レビューでは、DNAメチル化、ヒストン修飾、非コードmiRNAを含む様々なエピジェネティック修飾の変化と統合失調症およびその他の精神病性障害との間に中程度の関連性が確認されている。
初期の研究では、個々の候補遺伝子のプロモーターにおけるDNAメチル化変化の役割を分析した。統合失調症において、発達期および成人脳で重要な役割を果たす細胞外マトリックスタンパク質を코딩するリーリン遺伝子(RELN)の発現が、そのプロモーターの過メチル化により下方制御されることを示すいくつかの証拠がある。同様の観察により、GABA合成に必要な酵素をコードするグルタミン酸デカルボキシラーゼ67(GAD1)遺伝子のプロモーターが統合失調症と関連していることが判明した。さらに、GAD1プロモーターでは、H3K4me3の濃縮低下と抑制性マークであるH3K27me3の過剰も観察されている。統合失調症におけるGAD1調節異常の潜在的重要性を強調しているのは、GAD1とGABAが統合失調症で低レベルで発現するだけでなく、GAD1の変異が小児期発症統合失調症に関与し、GAD自己抗体症候群が典型的に統合失調症として現れるという確立された知見である。
さらに、クロマチン修飾因子として知られるヒストン修飾に関与する遺伝子の変異や構造変異は、様々な神経発達障害や統合失調症と関連している。例えば、ヒストンメチル化を仲介するタンパク質複合体の一部であるSETD1A遺伝子の機能喪失変異は、統合失調症のリスクを大幅に増加させる。SETD1Aのヘテロ接合性機能喪失変異を有するマウス系統は、皮質結合性とワーキングメモリーの欠陥を示している。さらに、マウス脳でSETD1Aによって推定制御される遺伝子は、発達期皮質で濃縮された神経発達遺伝子に濃縮されていた。これらの単一遺伝子型統合失調症は稀である可能性が高いが、統合失調症関連遺伝的変異の統合解析では、統合失調症の神経生物学へのクロマチン制御因子のより大きな寄与が示唆されている。
最近では、ゲノムワイドスケールで精神疾患のエピジェネティック機序の可能な役割を探る研究が行われている。例えば、正常脳発達中に観察されるDNAメチル化差異とゲノムワイド関連研究における統合失調症関連リスク遺伝子座を統合することにより、Jaffeらは、神経型の出生前サンプルと出生後サンプル間で全体的なCpGメチル化が異なることを観察した。出生前に過メチル化されたCpG部位は統合失調症リスク遺伝子座に濃縮されていた。しかし、疾患発症年齢(約25歳)でのリスクの有意な濃縮は観察されなかった。対照群と統合失調症成人前頭前皮質間のDNAメチル化プロファイルの直接比較により、約2,100のCpG遺伝子座が差次的にメチル化されていることが判明した。これらの遺伝子座は統合失調症リスク対立遺伝子に濃縮されていなかったが、再び神経型皮質での出生前後の変化を示した。
将来の方向性
神経精神疾患は、発達過程におけるゲノムの広範な調節異常に一部起因している。転写産物データは、複雑な精神疾患および神経発達障害において、多くの遺伝子の破綻が特定の細胞型と生物学的プロセスに収束して疾患表現型を生み出すことを示唆している。したがって、空間的・時間的に制御された遺伝子発現の選択的機能障害が、神経疾患や精神疾患における発症年齢と影響を受ける神経回路の違いを説明する可能性がある(図1.3-1参照)。しかし、疾患機序をさらに解明し、遺伝的・体細胞変異がどのように疾患を引き起こすかを決定し、より合理的な治療戦略を開発するためには、多くの研究が行われる必要がある。
転写産物の変化が障害の原因または結果を反映している可能性があることを明確にすることが重要である。これは、子宮内脳発達の破綻と関連するASDや統合失調症の成人患者からの死後脳サンプルにおいて最も顕著である。因果関係の方向を推測する一つの方法は、疾患ペアリング間の遺伝的相関とそれに対応する転写産物の重複を比較することであり、このアプローチによって神経精神疾患における遺伝子発現パターンの主要成分が根底にある遺伝的変異と結合した生物学的プロセスを反映していることが確認されている。別のアプローチは、疾患関連変異の大部分を占める遺伝子の非コード領域の変異に焦点を当てることである。具体的には、関連するCRE(シス調節エレメント)の時空間活性を特徴づけ、それらに関連する遺伝子を同定することで、疾患関連変異を転写産物変化に直接結びつけることができる。大規模並列レポーターアッセイ(MPRA)や量的形質遺伝子座(QTL)マッピングなどの現代的な実験アプローチや計算アプローチが現在、CREをより詳細に特徴づけ、特定の遺伝子と関連付けるために利用されている。
発達期ヒト脳のより包括的な転写産物、エピゲノム、制御マップを含む多次元ゲノムデータは、精神疾患などの複雑な疾患の病態生理学の現在の理解を大幅に進歩させるであろう。これらのデータセットは、いずれ診断やより標的化された治療パラダイムに有用な疾患および疾患サブタイプの遺伝的・分子表現型を明らかにする可能性がある。ゲノムおよび転写産物データの生成に加えて、病理学的、神経画像、行動学的、臨床的研究との統合解析が精神疾患の完全な理解にとって重要であろう。
FURTHER READINGS
BRAIN Initiative Cell Census Network (BICCN). A multimodal cell census and atlas of the mammalian primary motor cortex. Nature.
2021;598(7879):86-102.
Castelbaum L, Sylvester CM, Zhang Y, Yu Q, Constantino JN. On the nature of monozygotic twin concordance and discordance for autistic
trait severity: A quantitative analysis. Behav Genet. 2020;50(4):263-272.
Eze UC, Bhaduri A, Haeussler M, Nowakowski TJ, Kriegstein AR. Single-cell atlas of early human brain development highlights heterogen-
eity of human neuroepithelial cells and early radial glia. Nat Neurosci. 2021;24:584-594.
Gandal MJ, Haney JR, Parikshak NN, et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic over-
lap. Science. 2018;359(6376):693-697.
Hauberg ME, Creus-Muncunill J, Bendl J, et al. Common schizophrenia risk variants are enriched in open chromatin regions of human
glutamatergic neurons. Nat Commun. 2020;11:1-16.
Jeong H, Mendizabal I, Berto S, et al. Evolution of DNA methylation in the human brain. Nat Commun. 2021;12:2021.
Johnson MB, Kawasawa YI, Mason CE, et al. Functional and evolutionary insights into human brain development through global tran-
scriptome analysis. Neuron. 2009;62:494-509.
Kang HJ, Kawasawa YI, Cheng F, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483-489.
*Kelley KW, Paşca SP. Human brain organogenesis: Toward a cellular understanding of development and disease. Cell. 2022;185:42-61.
Kim MH, Kim IB, Lee J, et al. Low-level brain somatic mutations are implicated in schizophrenia. Biol Psychiatry. 2021;90(1):35-46.
Kwan KY, Lam MM, Johnson MB, et al. Species-dependent posttranscriptional regulation of NOS1 by FMRP in the developing cerebral
cortex. Cell. 2012;149:899-911.
Li M, Santpere G, Imamura Kawasawa Y, et al. Integrative functional genomic analysis of human brain development and neuropsychi-
atric risks. Science. 2018;362(6420):eaat7615.
Markenscoff-Papadimitriou E, Whalen S, Przytycki P, et al. A chromatin accessibility atlas of the developing human telencephalon. Cell.
2020;182:754-769.e18.
Miller JA, Ding SL, Sunkin SM, et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508:199-206.
Mukai J, Cannavò E, Crabtree GW, et al. Recapitulation and reversal of schizophrenia-related phenotypes in Setd1a-deficient mice. Neuron.
2019;104:471-487.e12.
Myers SM, Challman TD, Bernier R, et al. Insufficient evidence for “autism-specific” genes. Am J Hum Genet. 2020;106(5):587-595.
O’Leary DD, Chou SJ, Sahara S. Area patterning of the mammalian cortex. Neuron. 2007;56:252-269.
Olmos-Serrano JL, Kang HJ, Tyler WA, et al. Down syndrome developmental brain transcriptome reveals defective oligodendrocyte
differentiation and myelination. Neuron. 2016;89(6):1208-1222.
Parikshak NN, Luo R, Zhang A, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in aut-
ism. Cell. 2013;155:1008-1021.
Shibata M, Pattabiraman K, Lorente-Galdos B, et al. Regulation of prefrontal patterning and connectivity by retinoic acid. Nature.
2021;598:483-488.
Shibata M, Pattabiraman K, Muchnik SK, et al. Hominini-specific regulation of CBLN2 increases prefrontal spinogenesis. Nat.
2021;598:489-494.
Shim S, Kwan KY, Li M, Lefebvre V, Sestan N. Cis-regulatory control of corticospinal system development and evolution. Nature.
2012;486:74-79.
*Silbereis John C, Pochareddy S, Zhu Y, Li M, Sestan N. The cellular and molecular landscapes of the developing human central nervous
system. Neuron. 2016;89:248-268.
Stoyanova E, Riad M, Rao A, Heintz N. 5-Hydroxymethylcytosine-mediated active demethylation is required for mammalian neuronal
differentiation and function. Elife. 2021;10:e66973.
Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Gen.
2012;13:537-551.
*Tebbenkamp AT, Willsey AJ, State MW, Sestan N. The developmental transcriptome of the human brain: implications for neurodevelop-
mental disorders. Curr Opin Neurol. 2014;27:149-156.
Werling DM, Pochareddy S, Choi J, et al. Whole-genome and RNA sequencing reveal variation and transcriptomic coordination in the
developing human prefrontal cortex. Cell Rep. 2020;31:107489.
Willsey AJ, Sanders SJ, Li M, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis
of autism. Cell. 2013;155:997-1007.
Won H, Huang J, Opland CK, Hartl CL, Geschwind DH. Human evolved regulatory elements modulate genes involved in cortical expan-
sion and neurodevelopmental disease susceptibility. Nat Commun. 2019;10(1):2396.
Ziffra RS, Kim CN, Ross JM, et al. Single-cell epigenomics reveals mechanisms of human cortical development. Nature. 2021;598:205-
213.
