
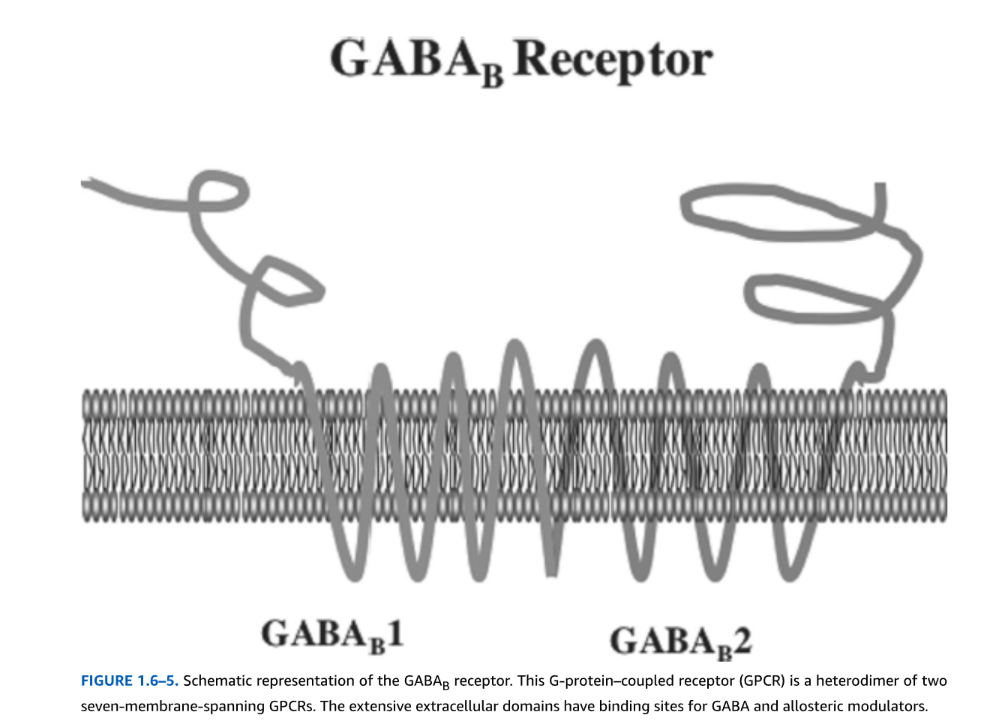
図1.6-5. GABAB受容体の概略図
このGタンパク質共役型受容体(GPCR)は、7つの膜貫通GPCRのヘテロ二量体です。広範な細胞外ドメインには、GABAおよびアロステリックモジュレーターの結合部位があります。
ナルコレプシーの治療薬として承認されている**γ-ヒドロキシ酪酸(GHB)**は、急速に深い睡眠を誘発するため、「デートレイプドラッグ」として誤用されてきました。脳内にはGHBの高親和性結合部位が報告されていますが、外因性GHBの鎮静作用および催眠作用はGABAB受容体拮抗薬によって予防できます。さらに、GABAB-/-マウスにGHBを投与してもGHBの行動効果は観察されず、GHBの作用部位はGHB受容体と呼ばれるものではなく、GABAB受容体である可能性が高いことを示しています。
神経伝達物質としてのグリシン
グリシンは主に脳幹と脊髄に存在する抑制性神経伝達物質ですが、視床、皮質、海馬におけるグリシン受容体サブユニットの発現は、より広範な役割を示唆しています。グリシンは非必須アミノ酸であり、脳内でセリンヒドロキシメチルトランスフェラーゼによってL-セリンから合成されます。グリシンは、H+依存性小胞抑制性アミノ酸トランスポーター(VIAATまたはVGAT)によってシナプス小胞内に濃縮され、このトランスポーターはGABAも輸送します。グリシンのシナプス作用の終結は、**グリシントランスポーターII(GlyT2)**によるシナプス前終末への再取り込みによって行われます。GlyT2はアストロサイトに発現し、NMDA受容体機能を調節するGlyT1とは全く異なります。
グリシンの抑制作用は、β-アラニン、タウリン、L-アラニン、L-セリン、プロリンにも応答しますが、GABAには応答しないリガンド依存性塩化物チャネルによって媒介されます。グリシン受容体の典型的な拮抗薬は植物アルカロイドのストリキニーネです。この受容体は、[3H]ストリキニーネのシナプス膜への特異的結合によって最初に同定されました。[3H]グリシン自体は2つの部位に結合します。1つはストリキニーネによって置換され、グリシンA受容体を表すもので、もう1つはストリキニーネに非感受性で、NMDA受容体上のGMSを表すグリシンB受容体と指定されています。
グリシンA受容体は、中心の孔を囲む5つのサブユニットからなる約250 kDaの巨大分子複合体です。高い相同性を持つ2つのサブユニットが存在します。それは48 kDaのαサブユニットと58 kDaのβサブユニットです。これらのサブユニットは、脂質二重層をαヘリックスとして貫通する4つの疎水性ドメインを持つ、このイオンチャネルファミリーの他のメンバーと構造的類似性を示します。グリシンとストリキニーネの両方の結合部位はαサブユニットに位置しています。αサブユニットをコードする遺伝子は4つありますが、βサブユニットをコードする遺伝子は1つだけです。興味深いことに、βサブユニットは、[3H]ストリキニーネ結合またはαサブユニット発現を示さない脳の吻側領域でかなり豊富に発現しています。
遺伝性驚愕病は、グリシン作動性シナプスの構成要素をコードする遺伝子の変異に起因する疾患です。これは乳児期の硬直と過剰な驚愕反射を特徴とし、成熟とともに軽減します。驚愕病を引き起こす変異は、グリシン受容体のαサブユニット(GLRA1)およびβサブユニット(GLRB)だけでなく、GlyT2(SLC6A5)でも報告されています。経口クロナゼパムによる治療は通常効果的な治療法です。
アミノ酸神経伝達物質の神経精神医学的示唆
統合失調症
死後、薬理学、および遺伝学研究から蓄積される証拠は、統合失調症の病態生理学の焦点をドーパミンからグルタミン酸とGABAへとシフトさせています。実際、過去60年間、様々な構造と副作用プロファイルを持つドーパミンD2受容体拮抗薬が統合失調症の主要な治療法として使用されてきましたが、治療された患者の3分の2以上が依然として著しい障害を抱えています。40年以上前に行われた死後研究では、統合失調症患者の大脳皮質においてGAD活性の低下が示されました。免疫細胞化学および遺伝子発現技術の出現により、統合失調症におけるGABA作動性欠損をより正確に定義することが可能になりました。皮質中間層のパルブアルブミン陽性GABA作動性介在ニューロンが病理の大部分を担っており、これにはGAD67、パルブアルブミン、およびGABAトランスポーター(GAT)の発現低下が含まれます。オートラジオグラフィーまたは抗体によって測定されるGABA受容体がアップレギュレートされているという発見は、これらの変化がシナプス前GABA作動性ニューロンの機能低下を反映しているという理論を支持しています。シャンデリア細胞を含むこれらの特定のGABA作動性介在ニューロンは、皮質の錐体細胞に対する負のフィードバック抑制において重要な役割を果たしています。
NMDA受容体の機能低下が統合失調症の病因因子であるという理論は、最初にPCPおよび関連するNMDA受容体拮抗薬である解離性麻酔薬が、統合失調症と区別できない症候群を引き起こすという観察から生じました。解離性麻酔薬は、意識の明らかなレベルを低下させることなく、新しい記憶の獲得を妨げるため、そのように名付けられています。実際、実験室条件下では、別の解離性麻酔薬であるケタミンの低用量注入は、明確な意識下で統合失調症に関連する陽性症状、陰性症状、および特定の認知機能障害を引き起こす可能性があります。その後の研究では、低用量ケタミンが、統合失調症で観察されるようなアンフェタミン誘発性皮質下ドーパミン放出の増強、ならびに異常な皮質事象関連電位(ERP)および実験動物におけるプレパルス抑制の障害も引き起こす可能性があることが示されました。
統合失調症の初期の遺伝子研究は、「候補」遺伝子の偏った選択と被験者数の少なさによって制限され、矛盾した結果をもたらしました。2014年、約4万人の統合失調症患者と10万人の対照者を比較した大規模なGWASは、GWASに固有の多重比較によって要求されるゲノムワイドな有意水準(5 × 10^-8)を達成した、ゲノム上の108の部位(一塩基多型[SNP])を特定しました(図1.6-6)。最近、これは統合失調症患者7万人と対照者23.6万人に拡大されました。明確なパターンが出現しました。リスク関連SNPのほとんどはゲノムのコーディング領域に局在しませんが、おそらく遺伝子発現またはRNAスプライシングを変化させることによって作用すると考えられています。注目すべきは、いずれも統合失調症の帰属可能なリスクに5%以上寄与するものはなく、統合失調症が糖尿病のような複雑な遺伝学的疾患であり、複数の小さな影響を持つリスク遺伝子が環境と相互作用して表現型を生み出すことを示唆しています。
3つの主要なリスク遺伝子カテゴリが特定されました。それは、主要組織適合性複合体(MHC)遺伝子、神経伝達関連遺伝子、および発生関連遺伝子です。SRR(セリンラセマーゼ、SRをコードする遺伝子)を含む約1ダースのリスク遺伝子はNMDARから2度以内の分離距離にあり、さらに1ダース以上の遺伝子が電位依存性Ca2+チャネルなど、グルタミン酸シグナル伝達に関与しています。マウスにおけるSRの遺伝的非活性化は、統合失調症の神経化学的変化、シナプススパインの消失などの神経構造欠損、EEG ERPsの障害などの電気生理学的異常、認知機能、ならびに精神病と一致する線条体ドーパミン放出の増加など、多くの特徴を共有する表現型をもたらします。双極性障害におけるGWASは統合失調症ほど強力ではありませんが、統合失調症と双極性障害の間で共有されるリスク遺伝子が特定されており、電位依存性カルシウムチャネルCACNA1CやNMDA受容体サブユニットGRIN2Aを含むグルタミン酸作動性神経伝達が双極性障害の病態生理に関与していることが示唆されています。
統合失調症患者におけるNMDA受容体のGMSに対するアゴニストを用いたいくつかの臨床試験が、抗精神病薬との併用治療を受けている患者を対象に実施されました。検証された仮説は、NMDA受容体機能を増強することが、抗精神病薬の影響を受けない障害の側面である陰性症状を軽減し、認知機能を改善するというものでした。NMDA受容体GMSに対するアゴニストのプラセボ対照試験の結果のメタアナリシスは、うつ病、陰性症状、認知機能障害、さらには陽性症状の非常に有意な減少を明らかにしました。しかし、異なる選択基準を用いた他のメタアナリシスは、説得力に欠ける結果を報告しており、GMSにおけるこれらの弱いアゴニストは臨床的に関連性がある可能性は低いが、「原理の証明」には寄与することを示しています。
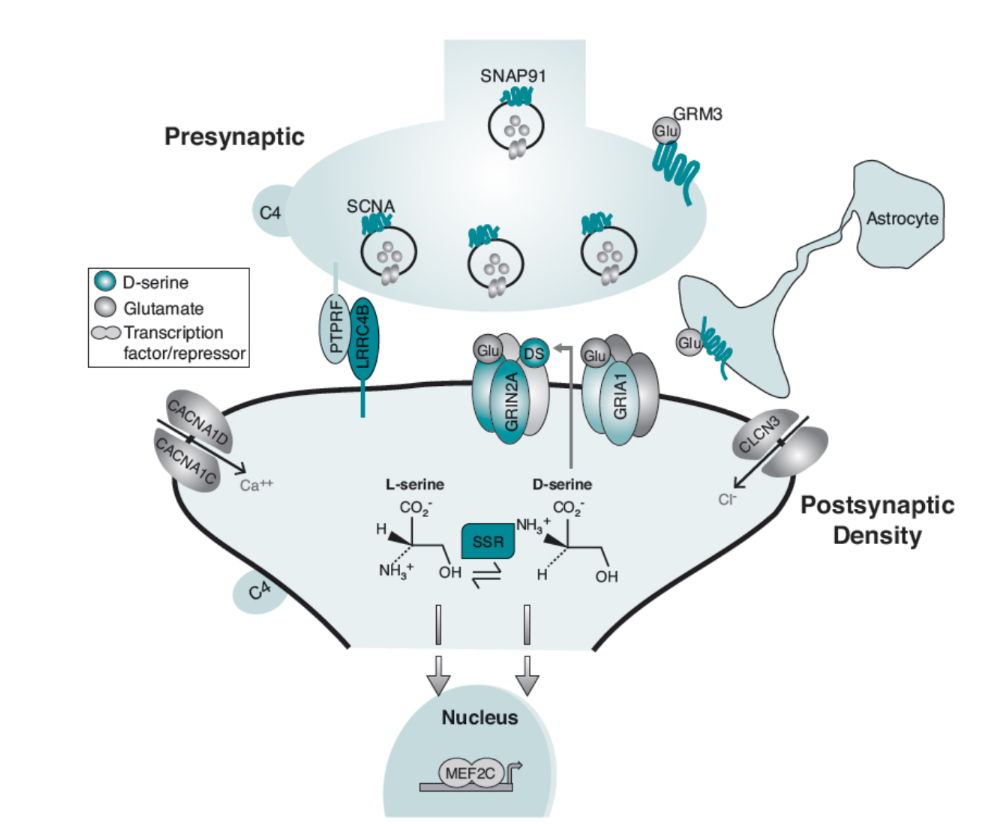
図1.6-6. 統合失調症の遺伝的リスクはグルタミン酸作動性シナプスに収束する。
統合失調症患者と対照者合わせて約20万人を対象としたゲノムワイド関連研究(GWAS)は、統合失調症のリスクとゲノムワイドに有意な関連(5 × 10^-8)を示す100以上のSNPを特定しました。関連するリスク遺伝子は、統合失調症の病態におけるグルタミン酸作動性シナプスの重要な役割を示しています。ここに示されている選択された統合失調症リスク遺伝子は、グルタミン酸作動性シナプス機能のあらゆる側面が統合失調症に関連していることを示しています。シナプス前終末では、SNCA(α-シヌクレイン)やSNAP91(シナプトソーム関連タンパク質91)などの遺伝子がシナプス小胞の輸送を調節します。PTPRF(プロテインチロシンホスファターゼ受容体F型)やLRRC4B(ロイシンリッチリピート含有4B)などのトランスシナプス細胞接着分子は、シナプスシグナル伝達、確立、およびリモデリングを調節します。電位依存性カルシウムチャネルサブユニットCACNA1C、CACNA1D、CACNA1Gや塩化物イオンチャネルサブユニットCLCN3など、複数の非受容体イオンチャネルサブユニット遺伝子が統合失調症に関連しています。同様に、グルタミン酸受容体のサブユニット自体も、NMDA受容体サブユニットGRIN2A、AMPA受容体サブユニットGRIA1とGRIA3、代謝型グルタミン酸受容体GRM1とGRM3を含む統合失調症リスク遺伝子によってコードされています。SRRによってコードされるセリンラセマーゼ酵素は、L-セリンをNMDA受容体の主要な共アゴニストであるD-セリンに変換します。主要組織適合性複合体(MHC)遺伝子座内のC4AおよびC4B遺伝子によってコードされる補体成分C4は、青年期後期の興奮性シナプスの過剰な刈り込みと、統合失調症で観察される樹状突起スパインの密度低下に寄与します。最後に、転写因子MEF2Cを含む複数の核因子が、NMDA受容体を介した遺伝子発現調節グルタミン酸作動性神経伝達を調節します。(Coyle JT, Ruzicka WB, Balu DT. Fifty years of research on schizophrenia: The ascendance of the glutamatergic synapse. Am J Psychiatry 2020;177(12):1119-1128. Copyright @ 2020. American Psychiatric Association. All Rights Reserved.より許可を得て転載。)
はい、以下に翻訳します。
統合失調症におけるGABA作動性神経病理とNMDA受容体機能低下の関連性
最近の発見は、統合失調症におけるGABA作動性神経病理とNMDA受容体機能低下との間に連結性があることを示しています。ラットをNMDA受容体拮抗薬で慢性的に治療すると、GAD67、パルブアルブミン、GATのダウンレギュレーションが引き起こされます。GABA作動性ニューロンの中で最も感受性の高いサブポピュレーションは、錐体細胞の周細胞体および軸索丘に神経支配を供給する急速発火性介在ニューロンです。これらのNMDA受容体は、活動が少ないGABA作動性ニューロンや錐体細胞のNMDA受容体よりも、拮抗薬に対してはるかに敏感であると考えられます。これは、NMDA受容体機能の重要な特徴が膜の脱分極だからです。GABBR1、GABBR2、およびCALB2をコードする遺伝子を除けば、現在のGWASの結果は、GABA作動性機能に関連する他の遺伝子を有意なリスク遺伝子として見出しておらず、統合失調症における皮質GABA作動性欠損は、NMDA受容体機能の低下など、GABA作動性ニューロン機能に影響を与えるより近位の遺伝的欠陥の下流の結果である可能性を示唆しています。
微妙に減少したGABA作動性抑制は、グルタミン酸作動性錐体細胞出力の脱抑制をもたらします。この皮質抑制性フィードバックの劣化は、統合失調症における認知機能障害と陰性症状の原因である可能性が高く、脱抑制された錐体細胞出力はまた、皮質下ドーパミン放出の増加をもたらし、精神病を引き起こします。統合失調症の表現型を模倣するこれらの発見は、前脳における主要なNMDA受容体共アゴニストであるD-セリンを除去する統合失調症のリスク遺伝子であるSRRをサイレンシングすることによって再現されます。したがって、精神病は、大脳皮質における重要なグルタミン酸作動性-GABA作動性シナプス機能の障害の下流の結果であると考えられます。
不安とうつ病
GABA作動性機能不全は、不安障害、特にパニック障害、および大うつ病性障害(MDD)と関連しています。臨床的には、不安障害と気分障害の間にはかなりの併存疾患があります。MRSは、MDD患者の前頭前野、帯状皮質、後頭葉皮質におけるGABAの有意な減少を明らかにしています。MDDのエピソードにおける後頭葉皮質におけるGABAレベルの減少は、選択的セロトニン再取り込み阻害薬(SSRI)または電気けいれん療法(ECT)による効果的な治療によって正常化します。死後研究では、自殺したうつ病患者の大脳皮質においてGABAA受容体α1-およびβ3-サブユニットのアップレギュレーションが明らかにされており、GABA作動性神経伝達の減少と一致しています。MDD患者の血漿およびCSFの両方で、GABAA受容体モジュレーターである3つのα還元神経活性ステロイドのレベル低下が発見されています。SSRIによる効果的な治療は神経ステロイドレベルを増加させます。MRSは、パニック障害の投薬を受けている患者の前帯状回および基底核におけるGABAレベルの有意な減少を明らかにしています。PETスキャンは、パニック障害における島皮質の両側におけるベンゾジアゼピン受容体部位の非常に選択的な減少を明らかにしています。ゲノムワイドスクリーニングでは、GABAA受容体サブユニット遺伝子とパニック障害を含む領域である15qに有意な連鎖が示されています。
グルタミン酸作動性機能不全もまた、うつ病に関与しています。NMDA受容体拮抗薬は、強制水泳、尾懸垂、学習性無力感など、いくつかの動物モデルのうつ病において抗うつ効果を示します。ケタミンの単回注射は、ラットにおける行動的絶望の誘発から最大10日間保護効果をもたらします。抗うつ薬による慢性治療は、NMDA受容体サブユニットの発現を変化させ、GMS結合を減少させます。いくつかのプラセボ対照臨床試験では、ケタミンの単回投与が、特にSSRI/SNRI治療に反応しなかったMDD患者において、症状の急速で実質的かつ持続的な軽減をもたらすことが示されています。さらに、ケタミン投与は自殺念慮の急速かつ持続的な減少を引き起こします。2019年、ヤンセンは、うつ病および自殺念慮の治療のために鼻腔内投与されるケタミンのS異性体(エスケタミン)の使用についてFDAの承認を得ました。ケタミンの急速な抗うつ作用の正確なメカニズムについては議論がありますが、ほとんどの研究は、NMDA受容体の一時的なブロックが、脳由来神経栄養因子(BDNF)放出の急速な増加と、特に報酬経路におけるグルタミン酸作動性神経伝達の強化をもたらすという点に集約されています。
アルコール依存症
酩酊に関連する濃度におけるエタノールは、GABA作動性受容体機能の増強とNMDA受容体機能の減弱という二重の作用を持ちます。GABAA受容体への影響は、エタノールの抗不安作用と関連している可能性があります。エタノールの持続的な乱用と依存は、GABAA受容体のダウンレギュレーションとNMDA受容体のアップレギュレーションをもたらし、エタノールの急激な中止は、振戦せん妄を特徴とする過興奮状態を引き起こします。さらに、チアミン欠乏症の状況における過敏性NMDA受容体は、ウェルニッケ・コルサコフ症候群の興奮毒性神経変性に寄与する可能性があります。
**アカムプロサート(キャンプラル)は、ホモタウリンの誘導体であり、アルコール使用障害(AUD)におけるアルコール摂取、渇望、再発を軽減する薬剤として開発されました。臨床試験では中程度の有効性を示します。タウリンがGABAに類似しているため、アカムプロサートはGABAA受容体を介して作用すると考えられていましたが、電気生理学的研究ではこの仮説を裏付ける証拠はほとんど見つかりませんでした。その後の研究では、皮質スライスおよび組換えNMDA受容体におけるNMDA受容体応答を阻害することが示されました。しかし、アカムプロサートがNMDA受容体機能を変化させる正確なメカニズムは不明なままです。最近、アカムプロサートは、AUDの再発をより良く予防するために、オピエート受容体拮抗薬であるナルトレキソン(ビビトロル)**と組み合わせて使用されています。
胎児性アルコール症候群(FAS)は、知的障害の最も一般的な予防可能な原因です。胎児期のアルコール曝露に関連する小頭症は、NMDA受容体機能の阻害に起因するという説得力のある証拠が開発されています。これは、未成熟ニューロンの生存と分化にはNMDA受容体活性化が不可欠であるため、未成熟皮質における広範な神経細胞のアポトーシスをもたらします。
結論
過去70年間、精神病に対するドーパミン機能の低減、うつ病および不安障害に対する生体アミン取り込みの阻害、抗不安作用のためのGABAA受容体機能の増強という基本的なメカニズムから逸脱したFDA承認の精神科治療薬はごくわずかでした。しかし、最近、AUDに対するアカムプロサート、治療抵抗性うつ病に対するNMDA受容体拮抗薬であるエスケタミン、産後うつ病に対するGABAA受容体アロステリックモジュレーターであるブレキサノロンなど、アミノ酸神経伝達物質機能を変化させることによって作用するいくつかの新規の有効な治療法が開発されました。
十分に強力なGWASを可能にする遺伝子研究の拡大は、グルタミン酸作動性およびGABA作動性神経伝達に関連する精神疾患の経路に関与するリスク遺伝子を現在明らかにしています。重篤な精神疾患におけるGWASの拡大は、治療開発のための非効率な方法である偶然への歴史的な依存から逸脱し、合理的な薬剤開発の標的を提供します。これらの進歩は、リスク遺伝子経路に関連する症状領域に対処する薬剤がますます導入され、診断と治療に対するカテゴリ別のアプローチを侵食するため、学生と精神科医にとって真の課題を提示するでしょう。脳神経伝達におけるそれらの卓越した役割を考えると、グルタミン酸とGABA経路はこれらの発展において重要な役割を果たす可能性が高いです。
参考文献
Anderson KM, Collins MA, Kong R, et al. Convergent molecular, cellular, and cortical neuroimaging signatures of major depressive dis- order. Proc Natl Acad Sci U S A. 2020;117:25138-25149.
Benham RS, Engin E, Rudolph U. Diversity of neuronal inhibition: a path to novel treatments for neuropsychiatric disorders. JAMA Psych- iatry. 2014;71:91-93.
Berg EL, Copping NA, Rivera JK, et al. Developmental social communication deficits in the Shank3 rat model of Phelan-McDermid syn- drome and autism spectrum disorder.1 Autism Res. 2018;11:587-601.
Lüscher B, Möhler H. Brexanolone, a neurosteroid antidepressant, vindicates the GABAergic deficit hypothesis of depression and may foster resilience.2 F1000 Res. 2019;8:F1000Faculty Rev-751
Bode A, Lynch JW. The impact of human hyperekplexia mutations on glycine receptor structure and function. Mol Brain. 2014;7:2.
Cheng HY, McGuinness LA, Elbers RG, et al. Treatment interventions to maintain abstinence from alcohol in primary care: systematic review and network meta-analysis. BMJ. 2020;371:m3934.
Cooke SF, Bear MF. How the mechanisms of long-term synaptic potentiation and depression serve experience-dependent plasticity in primary visual cortex. Philos Trans R Soc Lond B Biol Sci. 2013;369:20130284.
Corssen G, Domino EF. Dissociative anesthesia: further pharmacologic studies and first clinical experience with the phencyclidine de- rivative CI-581. Anesth Analg. 1966;45(1):29-40.
Coyle JT, Ruzicka WB, Balu DT. Fifty years of research on schizophrenia: the ascendance of the glutamatergic synapse.3 Am J Psychiatry. 2020;177:1119-1128.
Dienel SJ, Lewis DA. Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol Dis. 2019;131:104208.
Dutertre S, Becker CM, Betz H. Inhibitory glycine receptors: an update. J Biol Chem. 2012;287:40216-40223.
Gould TD, Zarate CA Jr, Thompson SM. Molecular pharmacology and neurobiology of rapid-acting antidepressants. Annu Rev Pharmacol Toxicol. 2019;59:213-236.
Granger AJ, Nicoll RA. Expression mechanisms underlying long-term potentiation: a postsynaptic view, 10 years on. Philos Trans R Soc Lond B Biol Sci. 2013;369:2013-2036.
Groc L, Choquet D. Linking glutamate receptor movements and synapse function. Science. 2020;368(6496):eaay4631.
Jensen AA, Fahlke C, Bjørn-Yoshimoto WE, Bunch L. Excitatory amino acid transporters: recent insights into molecular mechanisms, novel modes of modulation and new therapeutic possibilities. Curr Opin Pharmacol. 2015;20:116-123.
Lebois LAM, Seligowski AV, Wolff JD, Hill SB, Ressler KJ. Augmentation of extinction and inhibitory learning in anxiety and trauma- related disorders.4 Annu Rev Clin Psychol. 2019;15:257-284.
Lisman JE, Coyle JT, Green RW, et al. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizo- phrenia. Trends Neurosci. 2008;31:234-242.
Lynch JW, Zhang Y, Talwar S, Estrada-Mondragon A. Glycine receptor drug discovery. Adv Pharmacol. 2017;79:225-253.
Michalon A, Bruns A, Risterucci C, et al. Chronic metabotropic glutamate receptor 5 inhibition corrects local alterations of brain activity and improves cognitive performance in fragile X mice.5 Biol Psychiatry. 2014;75:189-197.
Moghaddam B, Krystal JH. Capturing the angel in “angel dust”: twenty years of translational neuroscience studies of NMDA receptor antagonists in animals and humans.6 Schizophr Bull. 2012;38:942-949.
Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39-47.
Pinar C, Fontaine CJ, Triviño-Paredes J, et al. Revisiting the flip side: long-term depression of synaptic efficacy in the hippocampus. Neuro- sci Biobehav Rev. 2017;80:394-413.
Price RB, Shungu DC, Mao X, et al. Amino acid neurotransmitters assessed by proton magnetic resonance spectroscopy: relationship to treatment resistance in major depressive disorder. Biol Psychiatry. 2009;65:792-800.
Proctor WR, Diao L, Freund RK, Browning MD, Wu PH. Synaptic GABAergic and glutamatergic mechanisms underlying alcohol sensitiv- ity in mouse hippocampal neurons. J Physiol. 2006;575:145-159.
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421-427.
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Mapping genomic loci implicates genes and synaptic biology in schizophrenia.7 Nature. 2022;604:502-508
Senter RK, Ghoshal A, Walker AG, Xiang Z, Niswender CM, Conn PJ. The role of mGlu receptors in hippocampal plasticity deficits in neurological and psychiatric disorders: implications for allosteric modulators as novel therapeutic strategies. Curr Neuropharmacol. 2016;14:455-473.
Singewald N, Schmuckermair C, Whittle N, et al. Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol Ther. 2015;149:150-190.
Swick TJ. Swick TJ. Treatment paradigms for cataplexy in narcolepsy: past, present, and future. Nat Sci Sleep. 2015;7:159-169.
Yang Y, Paspalas CD, Jin LE, Picciotto MR, Arnsten AF, Wang M. Nicotinic a7 receptors enhance NMDA cognitive circuits in dorsolateral prefrontal cortex.8 Proc Natl Acad Sci U S A. 2013;110(29):12078-12083. doi:10.1073/pnas.1307849110. Epub 2013 Jul 1.
Zorumski CF, Paul SM, Covey DF, Mennerick S. Neurosteroids as novel antidepressants and anxiolytics: GABA-A receptors and beyond. Neurobiol Stress. 2019;11:100196.
