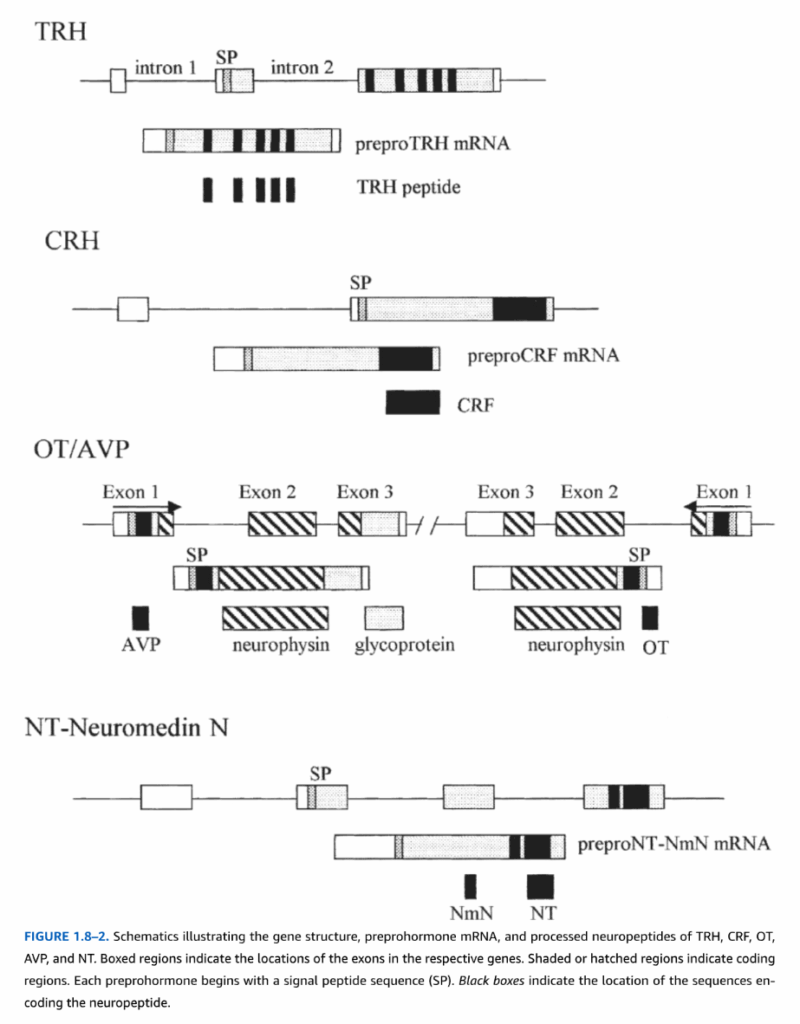
図1.8-2. TRH、CRF、OT、AVP、NTの遺伝子構造、プレプロホルモンmRNA、および処理された神経ペプチドを示す模式図
箱で囲まれた領域は、それぞれの遺伝子におけるエキソンの位置を示しています。陰影または斜線で示された領域は、コード領域を示しています。各プレプロホルモンは、シグナルペプチド配列(SP)から始まります。黒い箱は、神経ペプチドをコードする配列の位置を示しています。
多くの既知のペプチドは、プロホルモンから切断されたときに完全で生物学的に活性ですが、他の多くは追加の翻訳後修飾を受けます。特定のペプチドは、しばしばアミド化される代謝的にブロックされたカルボキシル末端を持ちます。プロホルモン配列中のグリシン残基は、しばしばアミド供与体として機能し、TRHの場合、分泌顆粒に含まれるモノオキシゲナーゼによって切断されます。TRHはN末端でさらに処理され、グルタミンがグルタミルシクラーゼによって環化されてピログルタミル部分を生成します。これらの変化は通常、分解への感受性を低下させるのに効果的であり、多くの場合、生物学的活性に必要とされます。TRHの場合がそうで、C末端のアミドがプロリンエンドペプチダーゼによって除去され、遊離酸構造が生成されると不活性になります。活性ペプチドの他の翻訳後修飾イベントには、グリコシル化、リン酸化、およびジスルフィド結合の形成が含まれ、これらはしばしば生物学的活性または輸送のいずれかに必要とされます。OT、バソプレシン、オレキシンAを含むいくつかの神経ペプチドは、1つ以上のシステイン-システインジスルフィド結合を含み、環状ペプチド構造を形成します(表1.8-2を参照)。最後に、神経ペプチドの多様性は、一次RNAの選択的スプライシングを介して異なるmRNAを生成し、最終的に著しく異なる作用を持つペプチド(例:カルシトニンとカルシトニン遺伝子関連ペプチド[CGRP])を生成することによって生じることができます。
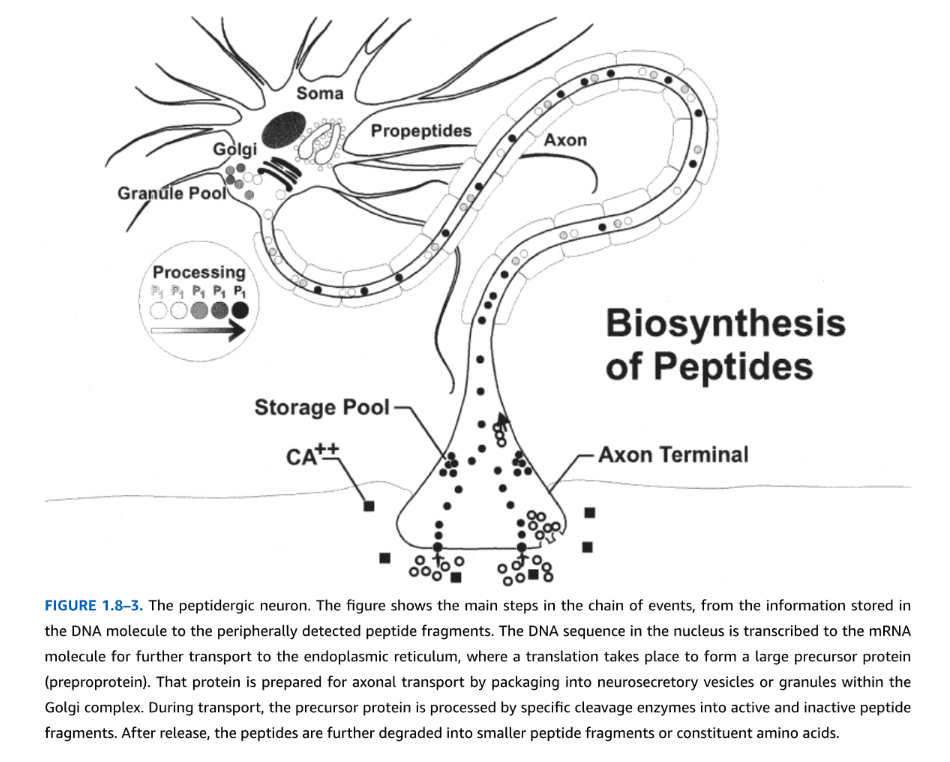
図1.8-3. ペプチド作動性ニューロン
図は、DNA分子に保存された情報から、末梢で検出されるペプチド断片までのイベントの主要なステップを示しています。核内のDNA配列はmRNA分子に転写され、さらに小胞体に輸送され、そこで翻訳が行われて大きな前駆体タンパク質(プレプロタンパク質)が形成されます。そのタンパク質は、ゴルジ複合体内で神経分泌小胞または顆粒にパッケージングされることにより、軸索輸送のために準備されます。輸送中に、前駆体タンパク質は特定の切断酵素によって活性および不活性ペプチド断片に処理されます。放出後、ペプチドはさらに小さなペプチド断片または構成アミノ酸に分解されます。
分布と調節
多くの神経ペプチドは元々下垂体や末梢組織から単離されましたが、大部分の神経ペプチドは脳全体に広く分布していることがわかっています。下垂体分泌の調節に関与するペプチドは、視床下部に集中しています。視床下部放出因子および放出抑制因子は、第三脳室に隣接する神経分泌ニューロンで産生され、正中隆起に投射して、そこでペプチドを視床下部-下垂体門脈循環系に放出します。これらのニューロンで産生されるペプチドは、しばしばそれらが調節する末梢ホルモンによるフィードバック調節を受けます。例えば、TRHは視床下部-下垂体-甲状腺(HPT)軸の活動を調節し、甲状腺ホルモンはTRH遺伝子発現を負にフィードバックします。しかし、神経ペプチドを発現するニューロンとその投射は、辺縁系構造、大脳皮質、中脳、後脳、脊髄を含む他の多くの脳領域で見られます。神経ペプチドは通常、排他的ではないにしても、他の神経ペプチドまたは古典的な神経伝達物質と共局在し、放出されます。古典的な神経伝達物質回路内の神経ペプチドの共局在は、1977年にソマトスタチンについて初めて記述され、これらのシステム間の相互作用を示唆しており、神経ペプチド(例:NPYとガラニン)によるモノアミン神経伝達物質(例:ノルエピネフリン)機能の調節は一般的でよく確立されています。実際、神経ペプチドは非ペプチド神経伝達物質と共伝達され、シナプス前細胞とシナプス後細胞の両方で神経伝達を調節する役割を果たします。
神経ペプチドシグナル伝達
神経ペプチドは、神経伝達物質、神経調節物質、または神経ホルモンとして作用する可能性があります。神経ペプチド神経伝達物質は通常、軸索終末からシナプスに放出され、そこで神経ペプチド受容体への結合がシナプス後膜電位を変化させ、細胞を脱分極または過分極させます。古典的な神経伝達物質の場合、これはしばしば電位依存性イオンチャネルの直接的な調節を伴います。対照的に、神経ペプチド神経調節物質および神経ホルモンは、標的細胞自体の発火に直接影響を与えるのではなく、セカンドメッセンジャー経路の調節を介して、他の神経伝達物質に対する細胞の応答を変化させる可能性があります。神経ペプチドの放出はシナプスや軸索終末に限定されず、軸索に沿った複数の部位、あるいは樹状突起からでも発生する可能性があります。神経ペプチドは、放出部位から神経ペプチド受容体を持つ標的細胞までかなりの距離を拡散し、そこで神経ホルモンとして作用し、ボリューム神経伝達と名付けられています。このようなパラクリン神経ペプチド放出は、グリア細胞などの脳内の非神経細胞集団からも発生する可能性があります。実際、脳内の神経ペプチドと神経ペプチド受容体の分布の間に不一致がある例は多数あります。神経ペプチドは、神経ペプチドを含むニューロンの電気的またはホルモン的刺激に応答して、顆粒のエキソサイトーシスによって放出されます。通常、神経ペプチドの放出は、ニューロンが高頻度で発火するか、バーストパターンで発火するときに発生します。刺激は細胞内カルシウム濃度の上昇をもたらし、これがペプチド顆粒と細胞膜の融合、およびペプチドの細胞外空間への放出につながります。
神経ペプチドの細胞シグナル伝達は、特定の神経ペプチド受容体を介して媒介されます。したがって、神経ペプチド受容体機能を理解することは、神経ペプチド生物学を理解するために不可欠です。神経ペプチド受容体は、他の神経伝達物質の受容体が享受してきた発見と特性評価の同じプロセスを経験してきました。神経ペプチド受容体の大部分は、モノアミン受容体と同じタンパク質ファミリーに属するGタンパク質共役型7回膜貫通型受容体(GPCR)です。各神経ペプチド受容体は通常、常にではないにしても、1種類のGタンパク質(例:Gs、Gi、Gq)と結合しています。受容体はホモダイマー化およびヘテロダイマー化も可能であり、単一の受容体集団を介して複数の作用様式を可能にします。ただし、このプロセス、その調節、および生理学的意味は、現在十分に理解されていません。受容体が相互作用するGタンパク質のサブタイプに応じて、受容体活性化は特定のセカンドメッセンジャー経路の刺激または阻害をもたらす可能性があります。最も一般的な種類の受容体シグナル伝達経路は、活性化されたGタンパク質がアデニル酸シクラーゼ(例:GsまたはGi)またはホスホリパーゼC(例:Gq)のいずれかの活動を調節することを含みます。アデニル酸シクラーゼの刺激は細胞内のcAMP産生を増加させ、ホスホリパーゼCの刺激はジアシルグリセロール(DAG)とイノシトール三リン酸(IP3)の増加をもたらします。これらの「セカンドメッセンジャー」は、細胞内カルシウム濃度の上昇、プロテインキナーゼの活性化、そして最終的には、遺伝子発現の変化、膜電位の変化、および/または細胞内顆粒脱顆粒を含む様々な細胞応答を引き起こします。
多くの神経ペプチドは、ペプチドに対する異なる親和性を持ち、異なるセカンドメッセンジャー経路を活性化する複数の異なる受容体サブタイプを介してその効果を発揮します。これらの受容体サブタイプは、通常、脳全体にわたって異なる分布をしています。さらに、多くの受容体は、複数の神経ペプチドによって調節される可能性があります。例えば、バソプレシン受容体にはV1a、V1b、V2の3つのサブタイプがあり、V1aとV1bは脳内で優勢であり、V2は腎臓に局在しています。これらの受容体サブタイプはそれぞれ、独自の組織分布を示し、異なるGタンパク質と相互作用し、異なるセカンドメッセンジャーシステムを活性化します。脳内では、OTとV1a受容体の分布に顕著な種差および個体差があり、これは種内および種間の社会行動の変動と関連付けられています。さらに、OTはバソプレシン受容体サブタイプを刺激する可能性があり、バソプレシンはOT受容体(OTR)を刺激する可能性があります。同様に、主要な2つのCRF受容体は脳内で異なる局在をしており、両方の受容体はCRFとウロコルチンIの両方によって調節される可能性があり、CRF機能における各受容体の相対的な役割を確定することは困難です。
すべての神経伝達物質ではないにしても、多くの神経伝達物質は、シナプスで少なくとも1つの他の神経伝達物質と共放出されるようです。例えば、ATPはすべてのニューロンタイプから共放出されるように見えますが、分子の作用は結合後細胞の生物学に応じて異なります。この共伝達は神経ペプチドにも確実に発生します。最初に解明されたものの1つであるNPYは、細胞内の同じ小胞からノルエピネフリンと共放出され、標的細胞に同時に作用します。NPYはまた、放出細胞にフィードバック阻害を介して作用し、シグナル伝達トーンを減少させることができます。この場合、NPYはシナプスで神経調節的な役割を果たし、信号強度を制御し、シャットオフスイッチとして機能するように見えます。ガラニンもノルエピネフリン放出ニューロンで同様の役割を果たします。
伝達物質と共伝達物質の差異放出(例えば、周波数変調放出による)、接合後および/または接合前標的化共伝達物質、相乗的または抑制的な共伝達物質など、他の種類の共伝達も発生する可能性があります。このようなシステムにおいて神経ペプチドを共伝達物質として認識することは重要です。なぜなら、共伝達は基礎となる疾患の病態生理を媒介または調節する可能性があるからです。NPYを再び例にとると、NPYは副腎でもNEと共伝達され、ストレスに応答してコルチゾールが放出されます。NPYは副腎内のNEシグナル伝達全体を抑制する役割を果たし、したがってストレス応答を抑制するように見えます。NPY受容体の活性化は、慢性ストレス障害や薬物乱用/アルコール依存症に関連するトラウマの一部を改善できるという理論が広く提唱されており、これらの状態ではストレス応答が長期にわたって活性化され、ストレスが再発や薬物探索行動において主要な役割を果たすと考えられています。
神経ペプチド受容体とリガンドのほとんどすべての遺伝子はクローン化され特性評価されていますが、「オーファン受容体」と呼ばれる一部の受容体は、現時点では未知の内因性リガンドを持っています。同様に、「コカインおよびアンフェタミン制御転写物」(CART)などの一部のリガンドは、まだ特定されていないGPCRを介してシグナル伝達すると考えられています。しかし、多くの努力が進行中であるため、既知のすべてのGPCRとリガンドは今後10年以内に「デオーファン化」される可能性が高いです。受容体のcDNAが単離されると、構造的および機能的研究のために精製された受容体タンパク質を生産するために使用できます。受容体構造内の特定のアミノ酸を変異させ、様々なアミノ酸置換を持つペプチドの相対結合親和性を決定することにより、リガンド-受容体相互作用の性質を解明し、受容体の結晶構造を通知することが可能です。この情報は、非ペプチド薬を含む受容体機能を特異的に調節する化合物の開発を促進し、より古典的な神経伝達物質が現在享受している方法でペプチドシステムを操作する能力につながるはずです。受容体をコードするcDNAの利用可能性は、受容体のmRNAを特異的に標的とするプローブの使用により、脳内の受容体産生細胞の神経解剖学的マッピングも可能にし、これはペプチドによって調節される神経回路を理解するために不可欠です。最後に、クローン化された受容体を手元に持つことで、標的遺伝子過剰発現や遺伝子ノックアウトなどのトランスジェニック技術を使用して、受容体機能をさらに解明することが可能です。ウイルス媒介RNAiおよびCRISPR技術は、特定の受容体集団の標的合成破壊を可能にし、研究者がこれらの受容体集団の生理学および行動への役割を検討できるようにします。ペプチド受容体Creトランスジェニック動物は、Cre依存性ベクターと組み合わせて、研究者が光遺伝学的および化学遺伝学的技術を使用して、受容体を発現する神経細胞集団の行動における役割を特定することを可能にします。
神経ペプチドホルモンの生物学的役割を決定する3つの要因は、(i)ペプチドの時間的解剖学的放出、(ii)神経ペプチド受容体と細胞内シグナル伝達経路の結合、および(iii)発現される受容体のサブタイプと異なる細胞集団および回路におけるその分布です。遺伝子研究は、受容体コード領域に隣接する調節配列が受容体の発現パターンを決定し、したがって神経ペプチドに対する生理学的および行動的応答を決定することを示しています。例えば、マウスとハタネズミは脳内のAVP受容体の局在が異なり、AVPに対する行動応答も異なります。しかし、ハタネズミのAVP受容体遺伝子とその隣接する調節配列を持つトランスジェニックマウスが作成されたとき、マウスはハタネズミに似たパターンで受容体を発現し、その後ハタネズミに似たAVPに対する行動応答を示しました。この研究は、神経ペプチド受容体遺伝子の調節領域における多型が、神経ペプチド機能に significant な違いをもたらし、神経刺激に対する個体差、そして最終的には精神疾患に関連する行動に significant な違いをもたらす可能性があることを示唆しています。多くの受容体遺伝子が、精神疾患との関連について遺伝子研究で検討されています。
歴史的に、特定の神経ペプチドシグナルを薬理学的にブロックできないことは、様々な行動および生理学的効果における内因性ペプチドの役割に関する研究を著しく妨げてきました。しかし、多くの神経ペプチド受容体では、現在選択的アゴニストと拮抗薬が利用可能であり、これらは受容体機能を検討する前臨床研究において非常に有益でした。上記のように、これらの化合物のほとんどはペプチド自体に由来し、したがって血液脳関門を通過しません。より最近では、多くの製薬会社、さらには学術研究室が、血液脳関門を透過し、神経ペプチドアゴニストまたは拮抗薬として作用する可能性のある非ペプチド性、脂溶性化合物を合成しています。これらの種類の化合物の開発は、ヒトの行動における神経ペプチド受容体機能の役割を理解するために不可欠であり、生きたヒト被験者における受容体分布を研究するための陽電子放出断層撮影(PET)放射性リガンドの開発にも有用である可能性があります。これらの化合物は、特定の精神疾患の治療における治療薬としての有望性も秘めています。
ペプチダーゼ
モノアミン神経伝達物質とは異なり、ペプチドはシナプス前神経終末による能動的な再取り込みによって不活性化されません。むしろ、放出されたペプチドは、ペプチダーゼと呼ばれる特定の酵素によって、より小さな断片、最終的には単一のアミノ酸に分解されます。これらの酵素は、シナプス後またはシナプス前の神経膜に結合しているか、細胞質および細胞外液中に溶解していることがあり、CNSだけでなく、末梢器官や血清にも広く分布しています。その結果、神経ペプチドは一般的に、放出されると数分程度の半減期を持ちます。ペプチダーゼにはいくつかの一般的なクラスがあり、各クラスにはいくつかの異なる酵素が含まれています。これらのクラスには、トリプシンやキモトリプシンなどの酵素を含むセリンエンドペプチダーゼ、ピログルタメートアミノペプチダーゼやカテプシンBおよびCなどのチオールペプチダーゼ、ペプシンやレニンなどの酸性プロテアーゼ、神経エンドペプチダーゼやアンジオテンシン変換酵素(ACE)などのメタロエンドペプチダーゼ、およびアミノペプチダーゼやエンケファリン変換酵素、カルボキシペプチダーゼAおよびBなどのメタロエキソペプチダーゼが含まれます。これらの分解酵素は、しばしば処理に使用されるものと同じですが、細胞内での局在が異なります。例としては、処理中にプロホルモン内の活性ペプチド配列を挟むジ塩基性アミノ酸残基を切断するカルボキシペプチダーゼBが挙げられます。NTのように、ペプチドが活性配列内にジ塩基性アミノ酸を含む場合、受容体での活性を低下させることもあります。ペプチダーゼは、活性に対して最適なpHと温度を持ち、様々な化学物質やキレート剤、またはペプチド鎖の脆弱な点でのアミノ酸置換によって阻害されることがあります。ペプチダーゼ活性または濃度の変化は、ペプチドのシナプス利用可能性の変化に寄与する可能性があり、ペプチダーゼレベルの調節は、受容体数、ペプチド合成、放出と同様に非常に精巧に制御されている可能性があります。活性な放出型ペプチドの切断は、通常、生物学的活性を終了させるか、大幅に減少させますが、部分的に代謝されたペプチドまたはその断片による部分的または完全な受容体活性化の例は豊富にあります。
ペプチダーゼは、神経ペプチド伝達物質の作用とシナプス利用可能性の統合と調節のためのさらなる潜在的な機会を提供します。現在のペプチダーゼ阻害剤は、様々なペプチダーゼを阻害する能力において比較的非特異的であるため、関連するペプチダーゼの薬理学的遮断によってペプチド濃度に影響を与えようとする試みはほとんどありませんでした。カプトプリルやリシノプリルなどのACE阻害剤はその一般性に対する例外の1つです。より新しい世代のペプチダーゼ阻害剤は、明確なペプチダーゼ特異性、およびおそらく領域特異性を持つものが開発され、最終的には内因性神経ペプチド濃度を真に優雅に操作できるようになることが期待されます。
薬物標的としての神経ペプチド系
神経ペプチド受容体が中枢神経系(CNS)において多くの調節的役割を果たすため、それらが魅力的な薬物標的となるのは当然のことです。精神疾患の治療のために神経ペプチド受容体を調節しようとする数多くの試みがなされており、いくつかの成功といくつかの注目すべき失敗がありました。治療薬の最初の、そして最も強力なクラス、特に末梢神経ペプチド受容体に関しては、神経ペプチド自体を投与することであり、おそらく薬物としての特性を向上させるためのわずかな修飾を加えることでした。しかし、精神疾患における問題は、大部分のペプチドが脳透過性ではないことです。脳透過性ペプチドを作成するか、あるいは中枢投与しようとする数多くの試みがありましたが、これらの努力は臨床的価値のあるものをほとんど生み出しませんでした。しかし、この分野は依然としてこのアプローチに焦点を当てており、遺伝子編集やその他の生物学的薬物アプローチにおける開発は、有用な結果をもたらす可能性を秘めています。
これらおよびその他の理由により、CNS神経ペプチド受容体システムを標的とするほとんどのアプローチは、低分子量(理想的には500 Da未満)の脳透過性分子の特定と開発に焦点を当てています。異なる神経ペプチド受容体を特異的かつ強力に標的とする多数の低分子が開発されています。これらの低分子は非常に多用途であり、非常に特異的な活性を持つように設計できます。一般的なアプローチは、受容体を活性化すること(アゴニスト)、受容体シグナル伝達を阻害すること(アンタゴニスト)、または内因性受容体活性を低下させること(逆アゴニスト)です。近年、より専門的なアプローチが開発されています。例えば、ポジティブアロステリックモジュレーター(PAM)低分子は、ペプチドが結合する部位(オーソステリック部位として知られる)ではないタンパク質上の部位(アロステリック部位として知られる)に結合することにより、神経ペプチドのシグナル伝達を増強し、同じ量の神経ペプチドで受容体により多くのシグナル伝達を開始させます。PAMアプローチは洗練されており、典型的なオーソステリックアゴニストが全身の受容体集団を活性化する可能性があるのと比較して、通常活性化されるはずの受容体のみを選択的に活性化するという潜在的な利点があります。同様のネガティブアロステリックモジュレーター(NAM)アプローチは、同じ量の神経ペプチドが存在する状況で受容体を介したシグナル伝達を減少させるために使用できます。最後に、近年、「バイアスのかかった」シグナル伝達薬の作成に大きな重点が置かれています。特定の受容体は、通常、正常な生理機能において活性化する複数の細胞内シグナル伝達経路を持っています。バイアスのかかった化合物は、他の経路よりも1つの経路を選択的に活性化または阻害することを目的としています。有名な例はμ-オピオイド受容体であり、β-アレスチンシグナル伝達システムを活性化しないオピオイド薬は、耐性が少なく、したがって依存症の可能性も低いと考えられています。これらの受容体を調節するための非常に専門的なアプローチは、現時点ではほとんど実験段階にありますが、そのような薬物を開発するための多くの努力が進行中です。未解決の主要な問題は、より堅牢なニューロン発火およびネットワークシステムの一般的にモジュレーターに過ぎない受容体シグナル伝達システムへの比較的軽微な調節が、肯定的な臨床結果をもたらすのに十分強力であるかどうかです。
神経ペプチドシステムが主に高度に調節的であるように見えるというこの問題は、神経ペプチドシステムのためのより伝統的な薬物探索が期待に応えられなかった理由でもあるかもしれません。神経ペプチド受容体薬の開発にはいくつかの顕著な失敗がありました。特に、2000年代半ばにほぼ同時期に起こった「ビッグスリー」の失敗は、神経ペプチド低分子薬が精神医学に有用であるかどうかを再考させるきっかけとなったようです。これらは、不安とうつ病に対するCRF受容体拮抗薬、痛みに対するNK1(SP)受容体拮抗薬、および肥満に対するNPY Y5受容体拮抗薬のフェーズIII臨床試験の失敗でした。有用性の可能性という疑問はほとんど未解決のままですが、アルツハイマー病(AD)治療薬の失敗など、他の神経科学分野でも同様の疑問と回答が現れています。ADの場合と同様に、薬物を開発する前に、正確なアプローチ、投与量、および作用機序をより明確に定義し、評価することが不可欠です。さらに、試験を開始する前に、疾患と臨床転帰についてより徹底的に検討する必要があります。明確な例は、全般性不安障害(GAD)に対して意味のある結果をもたらさなかったCRF受容体拮抗薬です。GADは、多くのDSM5障害と同様に、非常に多様です。CRF受容体拮抗薬のアプローチは、例えば、CRF活性の増加を促進する遺伝的および小児期の虐待歴を持つ外傷後ストレス障害(PTSD)患者など、より明確で豊かな患者のサブポピュレーションにおいて非常に効果的である可能性があります。すべての神経精神疾患において、臨床集団、臨床試験デザイン、試験結果、その他の要因は、成功のチャンスがあるならば、これまで以上に注意を払う必要があります。明確な例として、本章で後述するように、CRF過剰分泌が気分障害、不安障害、およびアルコール使用障害(AUD)の病態生理学的基礎となる可能性があるという継続的な仮説は、重要な前臨床および臨床研究によって裏付けられています。しかし、これらの障害を持つ個人の一部、おそらく20%から30%だけがCRF過剰分泌者であると予想されます。広範な診断カテゴリに基づいた臨床試験デザインは、これらの薬剤の価値を評価する際に莫大な統計的パワーを失うだけでなく、正常なCRF機能を持つ被験者に不都合な影響を引き起こす可能性があります。この分野は、これらの問題に概ね取り組んでおり、神経ペプチド受容体低分子臨床試験の開発の再燃が期待されています。
神経ペプチド生物学の代表としての特定の神経ペプチド
甲状腺刺激ホルモン放出ホルモン(TRH)
1969年、ピログルタミル-ヒスチジル-プロリルアミドトリペプチドであるTRH(表1.8-2参照)は、視床下部放出ホルモンとして最初に単離され特性評価されました。この研究は、アンドリュー・シャリー博士とロジャー・ギルマン博士に1977年のノーベル賞をもたらした部分的な成果でもあります。このホルモンの構造の発見は、視床下部から分泌されるペプチドホルモンが下垂体前葉からのホルモン分泌を調節するという決定的な実証につながりました。ヒトのTRH遺伝子は染色体3q13.3-q21に存在します。1ラットでは、2つのイントロン(非コード領域)によって分離された3つのエキソン(コード領域)から構成されます(図1.8-2参照)。最初のエキソンはTRHプレプロホルモンをコードするmRNAの5’非翻訳領域を含み、2番目のエキソンはSP配列と前駆体ペプチドの残りのアミノ末端の大部分を含み、3番目のエキソンはTRH前駆体配列の5つのコピー、カルボキシル末端領域、および3’非翻訳領域を含む残りの配列を含みます。遺伝子の5’側隣接領域、すなわちプロモーターは、グルココルチコイド受容体および甲状腺ホルモン受容体DNA結合部位に相同な配列を含み、コルチゾールによるこの遺伝子の調節および甲状腺ホルモンによる負のフィードバックのメカニズムを提供します。TRHの酵素的処理は、カルボキシペプチダーゼによる前駆体ペプチドの切断、C末端プロリンのアミド化、N末端グルタミンの環化から始まり、プロホルモン分子あたり5個のTRH分子を生成します。TRHはCNSに広く分布しており、TRH免疫反応性ニューロンは嗅球、嗅内皮質、海馬、拡張扁桃体、視床下部、および中脳構造に位置しています。ほとんどの神経ペプチドの場合と同様に、TRH受容体も7回膜貫通型Gタンパク質共役型受容体ファミリーのメンバーです。
視床下部のTRHニューロンは、神経終末を正中隆起に投射し、そこでTRHを視床下部-下垂体門脈系に放出し、それはアデノヒポフィシスに輸送され、TSHの全身循環への放出を引き起こします。TSHはその後、甲状腺から甲状腺ホルモンであるトリヨードチロニン(T3)とチロキシン(T4)の放出を刺激します。傍室核のTRHニューロンは甲状腺ホルモン受容体を含み、甲状腺ホルモン分泌の増加に応答してTRH遺伝子発現と合成の減少を示します。甲状腺切除後、正中隆起(甲状腺機能を促進しようとして放出が増加したため)ではTRH含有量が減少しましたが、視床下部の傍室核では減少しませんでした。この甲状腺ホルモンによるTRH合成ニューロンへの負のフィードバックは、最初にこの方法で実証されました。この効果は、外因性甲状腺ホルモン治療によって逆転させることができます。正常なラットへの外因性甲状腺ホルモン治療は、傍室核と視床下部の後核におけるTRH濃度を減少させます。TRHプレプロホルモンmRNAに対するプローブを使用したin situハイブリダイゼーション研究は、甲状腺切除14日後に傍室核においてTRH mRNAが増加することを示しました。甲状腺ホルモンがTRH mRNAを調節する能力は、HPT軸を活性化する他の刺激によって上書きされる可能性があります。その点に関して、寒冷への繰り返し曝露(正中隆起からTRHを放出する)は、甲状腺ホルモン濃度が同時に上昇しているにもかかわらず、傍室核におけるTRH mRNAレベルの増加を誘発します。HPT軸の異なるコミュニケーションレベルのさらなる証拠は、下垂体TRH受容体のmRNA産生を調節するTRHの能力、およびTRH濃度が甲状腺刺激ホルモン(TSH)分子のαサブユニットとβサブユニットの両方をコードするmRNAを調節する能力に見られます。さらに、TRH含有シナプスボタンが傍室核の内側および脳室周囲のサブディビジョンにおけるTRH含有細胞体と接触しているのが観察されており、これによりTRH放出の超短フィードバック調節の解剖学的証拠が提供されています。甲状腺ホルモンによる負のフィードバックは、視床下部のTRHニューロンに限定される可能性があります。なぜなら、甲状腺ホルモンによるTRH合成への負のフィードバックは、視床下部外TRHニューロンでは見つかっていないからです。
HPT軸機能の評価に十分なツール(すなわち、ラジオイムノアッセイ、合成ペプチド)が早期に利用可能になったこと、そして原発性甲状腺機能低下症が抑うつ症状と関連しているという観察が相まって、この軸が感情障害に関与していることについて広範な調査が確実に行われました。初期の研究により、TRHの視床下部および視床下部外分布が確立されました。TRHのこの視床下部外での存在は、TRHが神経伝達物質または神経調節物質として機能する可能性があるという憶測に迅速につながりました。実際、膨大な証拠がTRHのそのような役割を裏付けています。CNS内では、TRHはドーパミン、セロトニン、アセチルコリン、オピオイドなど、いくつかの異なる神経伝達物質を調節することが知られています。TRHは冬眠動物を覚醒させ、バルビツール酸塩やエタノールを含む様々なCNS抑制剤によって生成される行動反応や低体温症に対抗することが示されています。
TRHのCNS作用の可能性への関心は、PrangeらのHPT軸とうつ病に関する研究によって刺激されました。約40年前、甲状腺ホルモン、カテコールアミン、CNSのアドレナリン受容体間の多数の相互作用により、甲状腺機能が感情障害の病因と回復に不可欠であると仮説が立てられました。全体として、これらの研究は難治性うつ病における甲状腺機能不全の役割を示唆しており、難治性うつ病患者における甲状腺機能低下症および自己免疫性甲状腺炎の発生率の増加を示唆する臨床研究と一致しています。
HPT軸機能評価のための刺激剤としてのTRHの使用は、その単離と合成後、急速に発展しました。標準化されたTRH刺激試験の臨床的使用(負のフィードバック応答を測定する)は、主要な抑うつ症状を持つ甲状腺機能正常患者の約25%でTSH応答の鈍化を明らかにしました。これらのデータは広く確認されています。抑うつ患者で観察されるTSHの鈍化は、甲状腺ホルモン濃度の基礎血漿濃度などの甲状腺測定値がこれらの患者では一般的に正常範囲であるため、甲状腺機能亢進症による過剰な負のフィードバックの結果ではないようです。TSHの鈍化は、内因性TRHの正中隆起過剰分泌の結果として、下垂体TRH受容体のダウンレギュレーションを反映している可能性があります。実際、対照と比較して抑うつ患者のCSF TRH濃度が上昇しているという観察は、TRH過剰分泌の仮説を支持しますが、このトリペプチドのCNS起源を明確にはしません。実際、主要な抑うつ症状を持つ患者では、視床下部の傍室核におけるTRH mRNA発現は減少しています。しかし、変化したHPT軸が抑うつ症状の根底にある原因メカニズムであるのか、あるいは単に他の神経系における抑うつ関連の変化の二次的な影響であるのかは明確ではありません。最近、Cohenらは、血漿TSH濃度の軽度な増加(>3.5 mIU/mL)でさえ、厳密にはいわゆる「正常」範囲の上限ですが、抗うつ剤治療への反応が不良であることと関連していると報告しています。
副腎皮質刺激ホルモン放出因子とウロコルチン
1950年代には、下垂体抽出物中にCRFと呼ばれる因子が含まれており、生体内で下垂体前葉細胞からのACTH放出を刺激することが観察されました。約30年間の探索の後、Valeらは1981年にCRFを41アミノ酸ペプチドとして単離し、特性評価しました。ヒトのCRF遺伝子は染色体8q13に位置し、2つのエキソンから構成され、CRFプレプロホルモンは完全にエキソン2にコードされています(図1.8-2参照)。より最近、関連する神経ペプチドであるウロコルチン1、ウロコルチン2、ウロコルチン3が同定され、同様の遺伝子構造を共有しています。CRFはほとんどの種において主要な視床下部ACTH分泌促進因子であり、また、ウロコルチンとともに、ストレス要因に対する応答を全体的に調整するCNSネットワークにおいて視床下部外神経伝達物質/神経調節物質としても機能します。CRFとウロコルチンが、生物のストレスに対する内分泌、自律神経、免疫、および行動応答の統合において複雑な役割を果たすという仮説を裏付ける説得力のある証拠があります。
CRFは当初、視床下部-下垂体-副腎(HPA)軸の調節機能のために単離されましたが、脳全体に広く分布しています。視床下部の傍室核(PVN)は、下垂体前葉ACTH分泌に影響を与えるCRF含有細胞体の主要な部位です。これらのニューロンはPVNの小細胞領域に由来し、軸索終末を正中隆起に送ります。そこでCRFはストレス刺激に応答して視床下部-下垂体門脈系に放出されます。少数のPVNニューロンは脳幹および脊髄にも投射し、そこでストレス応答の自律神経側面を調節します。CRF含有ニューロンは、他の視床下部核、新皮質、拡張扁桃体、脳幹、および脊髄にも見られます。実験動物へのCRF中枢注入は、運動活動の増加、音響驚愕反応の増加、オープンフィールドでの探索行動の減少など、ストレス後に観察されるものと同様の生理学的変化および行動効果を生じさせます。
TRHと甲状腺ホルモンについて述べたのと同様に、PVNにおけるCRF遺伝子発現と含有量はグルココルチコイド(コルチゾール)によって負に影響され、様々なストレス要因によって正に調節されます。副腎摘出はPVNにおけるCRF mRNA発現の増加をもたらし、グルココルチコイド補充は用量依存的にCRF mRNA発現を減少させます。PVNへの影響とは対照的に、グルココルチコイドは扁桃体におけるCRF mRNA発現を減少させるのではなく増加させます。
CRFはまた、縫線核と青斑核(LC)にも見られます。これらはそれぞれ、前脳への主要なセロトニン作動性およびノルアドレナリン作動性投射の起源であり、うつ病と不安症の病態生理に役割を果たすと長い間仮説が立てられてきた回路です。CRFのCNSへの直接投与後に観察される不安の増加は、部分的にノルアドレナリン作動性活動の増加によって媒介されると仮説が立てられています。ストレスは、青斑核におけるCRF含有量の増加と、正中隆起におけるCRF濃度の減少(放出の増加と一致)を引き起こすことが示されています。他の研究では、CRF含有神経終末が青斑核のノルアドレナリン作動性ニューロンに作用し、それらのニューロンに外因性CRFを適用すると、それらの発火率が変化することが示されています。これらのノルアドレナリン作動性青斑核ニューロンの一部は、視床下部PVNに投射し、その入力がCRF合成と放出を増加させます。青斑核へのCRF注入が恐怖または不安行動を誘発するため、ストレスが青斑核ノルアドレナリン作動性ニューロンに終末するCRFニューロンを活性化し、それがPVNへの他の入力と共同して、ストレス誘発性の正中隆起からのCRF放出増加を刺激する可能性があると推測できます。近年追求されているもう1つの仮説は、重度のストレスが側坐核ドーパミンニューロンをCRFに非感受性にさせ、ドーパミン放出の減少と快楽欠如の増加をもたらすというものです。興味深いことに、人生の早期に母子分離に曝された成人動物(早期の有害な小児期の経験の動物モデルであり、非常に重度のストレス形態)は、LCにおけるCRF濃度の増加、CRF mRNA発現の増加、およびストレスに対するHPA軸応答の誇張を示します。関連して、ますます多くの証拠が、薬物乱用障害におけるストレス誘発性再発におけるCRFの役割を強く支持しています。
ウロコルチンの生理学的および行動的役割はあまり理解されていませんが、いくつかの研究はウロコルチン2と3が抗不安作用を持ち、ストレス応答を緩和する可能性があることを示唆しています。これにより、CRFとウロコルチンが反対に作用するという仮説が提唱されましたが、これは単純化しすぎである可能性が高いです。ウロコルチン1は主にエディンガー・ウェストファール核、外側オリーブ核、および視索上核で合成されます。ウロコルチン2は主に視床下部で合成され、ウロコルチン3の細胞体は拡張扁桃体、室傍核領域、および視前野に広く見られます。
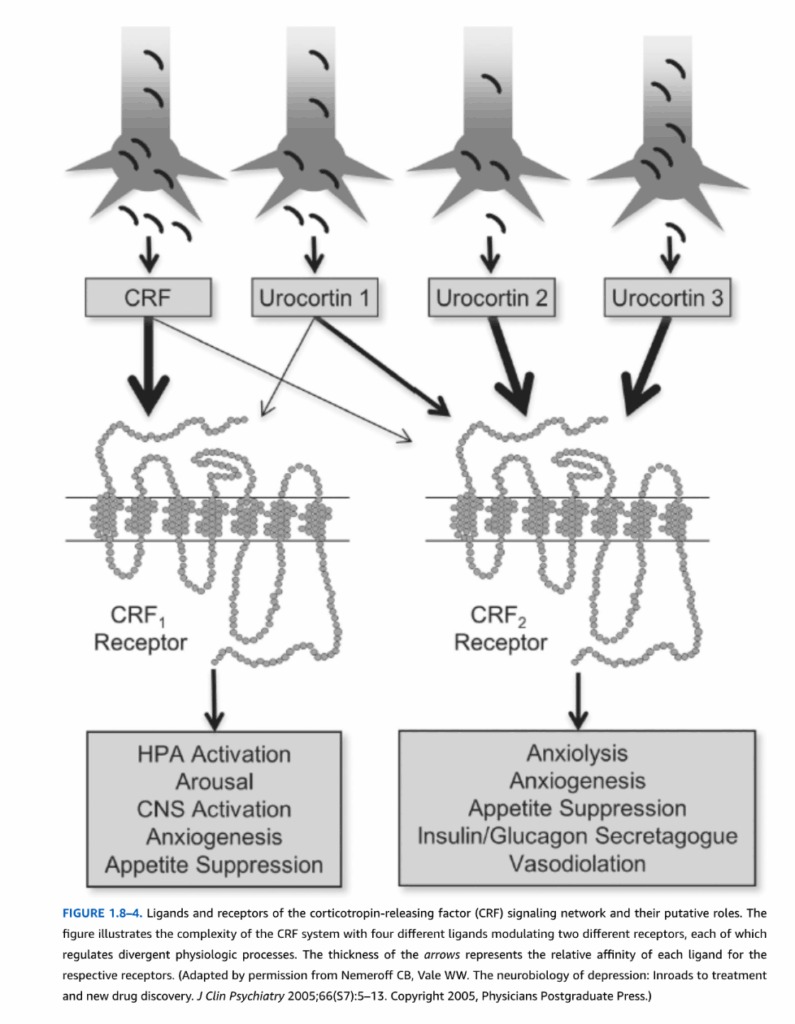
CRFシステムの複雑さは、CRFとウロコルチンの効果が少なくとも2つの受容体サブタイプ、CRF1受容体とCRF2受容体(図1.8-4)によって媒介されるという事実によってさらに増します。CRF1受容体は大脳皮質、小脳、内側中隔、下垂体前葉に豊富に発現していますが、CRF2受容体はげっ歯類では外側中隔、腹内側視床下部、脈絡叢に主に存在しますが、ヒトの皮質ではかなりの発現が見られます。CRF1受容体は、ストレス応答におけるCRFの効果を媒介する主要な受容体であるようです。CRF1受容体は、ウロコルチン1よりもCRFに対して4〜10倍高い親和性を持ち、他のウロコルチンに対しては非常に低い親和性を示します。対照的に、CRF2受容体はCRFと比較してウロコルチンに対して40倍高い親和性を持ちます。したがって、ウロコルチンはCRF2受容体の内因性リガンドであると提唱されていますが、その生理学的役割についてはほとんど知られていません。予想通り、CRF1受容体ノックアウトマウスは、不安様行動の減少、ストレス応答の障害、およびグルココルチコイドの負のフィードバックの欠如によるPVNにおけるCRF mRNA発現の上昇を示します。対照的に、CRF2受容体ノックアウトマウスは、不安様行動の増加およびストレスに対する過敏性を示します。
大うつ病におけるHPA軸の過活動は、生物精神医学における最も一貫した所見の1つであり続けています。大うつ病で報告されているHPA軸の変化には、コルチゾール過剰分泌、デキサメタゾンによるコルチゾール分泌抑制抵抗性(負のフィードバックの指標)、静脈内CRF負荷に対するACTH応答の鈍化、デキサメタゾン/CRF併用試験におけるコルチゾール応答の増加、およびCSF CRF濃度の上昇が含まれます。大うつ病および他の感情障害におけるHPA軸調節不全の根底にある正確な病理学的メカニズムは未解明のままです。