
- 1.9 新規神経伝達物質
- 図1.9-1:作動薬、拮抗薬、部分作動薬、および逆作動薬
- 神経伝達物質としてのガス
- 図1.9-2:一酸化窒素の酵素的生成
- 図1.9-3:サイクリックGMP産生を介した一酸化窒素(NO)の神経伝達物質およびシグナル伝達機能
- 図1.9-4:S-ニトロシル化を介した一酸化窒素(NO)シグナル伝達
- 図1.9-5:NMDA受容体活性化後の一酸化窒素(NO)生成
- 一酸化炭素
- 図1.9-6:非典型的神経伝達物質である一酸化炭素の合成
- 一酸化炭素のその他のシグナル伝達の役割
- 硫化水素:最新のガス状メッセンジャー分子
- エンドカンナビノイド:マリファナから神経伝達へ
- 図1.9-7:植物由来および合成カンナビノイドの選択
- 図1.9-8:内因性カンナビノイド
- 図1.9-9:エンドカンナビノイド(アナンダミドと2-アラキドノイルグリセロール(2-AG))の逆行性神経伝達
1.9 新規神経伝達物質
トーマス・W・セドラック 医学博士、ビンデュ・D・ポール 博士、アダム・I・カプラン 医学博士
神経伝達物質は、あるニューロンから隣接するニューロンへの脱分極シグナルを増幅または抑制する化学物質です。神経伝達物質は通常、シナプス前ニューロンから放出され、シナプス間隙またはシナプスという小さな空間を越えて移動し、シナプス後ニューロンに作用します。神経伝達物質は、標的細胞の脱分極を促進する点で興奮性(例:グルタミン酸)である場合もあれば、過分極を促進する点で抑制性(例:ほとんどのGABA)である場合もあります。イオンチャネルである神経伝達物質受容体は、通常、ナトリウムまたはカルシウムを介して脱分極を、塩化物イオンを介して過分極を引き起こします。イオンチャネル受容体は、メタボトロピック受容体とは異なります。後者は、遺伝子発現を変化させたり、脱分極の傾向を調節したりするなど、標的細胞に複雑な影響を与えるセカンドメッセンジャーを産生します。
活動電位は、ニューロンの軸索を伝わり、神経伝達物質が特殊な小胞に貯蔵されている特殊な付属物であるシナプス前終末に到達する脱分極の波です。活動電位は膜内の電位依存性カルシウムチャネルを開き、細胞内カルシウムの増加を引き起こします。これにより、小胞がその内容物をシナプス間隙に放出し、シナプス後ニューロンの膜上の受容体に作用します。標的に結合する際、神経伝達物質は通常、完全作動薬または部分作動薬であり、治療薬は部分作動薬から完全作動薬、拮抗薬、または逆作動薬まで様々です(図1.9-1)。
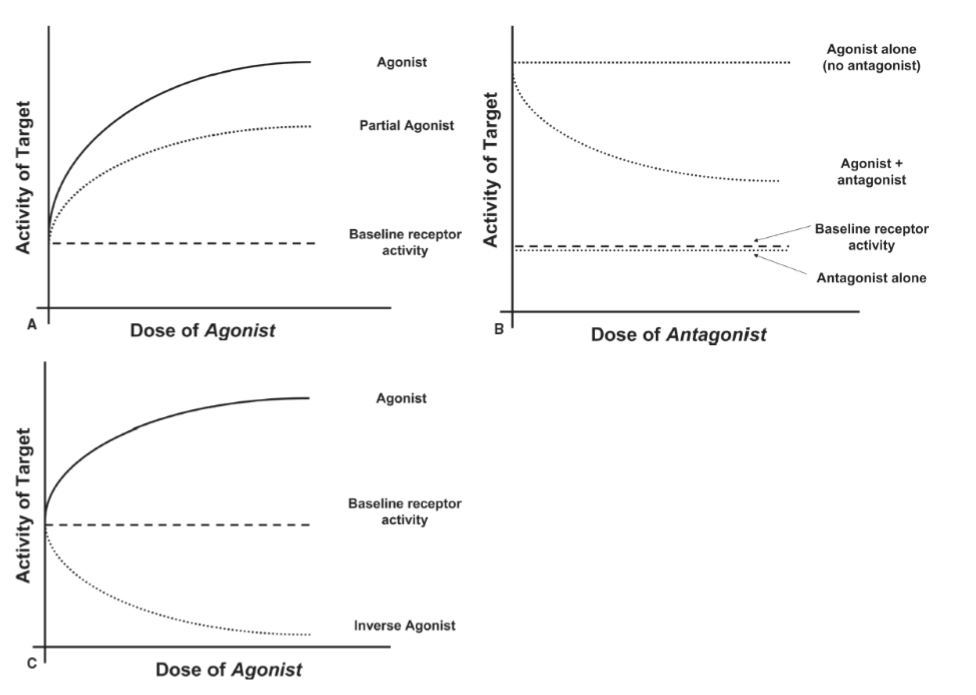
図1.9-1:作動薬、拮抗薬、部分作動薬、および逆作動薬
A:作動薬は、神経伝達物質受容体などの標的に結合し、受容体が飽和するまでその活動を増加させます。部分作動薬は、標的の活動を増加させますが、その最大レベルを下回るレベルに留まります。場合によっては、部分作動薬は完全作動薬である神経伝達物質と競合するため、神経伝達物質受容体の全体的な活動が減少します。まれに、作動薬と見なされていた薬物が、より強力な作動薬が発見された後に部分作動薬として再分類されることがあります。THCはかつてCB1受容体の完全作動薬と考えられていましたが、より強力な合成カンナビノイドであるCP55,940とWIN55,212-2の発見後、部分作動薬であることが判明しました。
B:拮抗薬は、受容体を活性化または抑制する内在的な活性を持ちません。多くの場合、結合部位で作動薬と競合することにより、作動薬の活動を阻害するよう作用します。
C:逆作動薬は、薬物がないベースラインレベルよりも低いレベルに標的の活動を抑制します。リモンナバンはCB1受容体逆作動薬です。カンナビノイド作動薬が存在しない場合でも、受容体のベースライン活性をブロックすることができます。
神経伝達物質が何であり、何でないかという定義は、数十年にわたって変化してきました。最初に発見された神経伝達物質は小さな化学物質で、最初はアセチルコリン、その後はセロトニン、ドーパミン、ノルエピネフリン、エピネフリン、ヒスタミンなどの生体アミンでした。その後、エンケファリンがオピオイド受容体に作用するように、アミノ酸やペプチドが神経伝達物質として機能することが判明しました。1990年代には、カンナビノイド受容体に作用する神経伝達物質が細胞脂質に由来することが明らかになりました。さらに、一酸化窒素というガスでさえも神経伝達物質となり、膜受容体を迂回してシナプス後ニューロン内で直接作用することがわかりました。
神経伝達物質としてのガス
一酸化窒素
ガスが神経伝達物質として機能することが発見されたことにより、ニューロン間に非常に非典型的なシグナル伝達様式が存在することが明らかになりました。1990年代初頭、一酸化窒素は神経伝達物質機能が帰属された最初のガスであり、いくつかの理由で非典型的な神経伝達物質であることが判明しました。第一に、それはシナプス小胞に貯蔵されたり、そこから放出されたりしないことでした。それは小さなガスであり、細胞膜を自由に拡散して標的ニューロン内に入ることができました。第二に、その標的は標的ニューロン表面の特定の受容体ではなく、活動が直接調節されうる細胞内タンパク質でした。一酸化窒素には、シナプスからそれを取り除く再取り込み機構もありません。酵素的失活が存在すると仮定されていますが、一酸化窒素は数秒という短い半減期によって制限されます。
一酸化窒素は、当初、マクロファージから放出される殺菌化合物として、また血管拡張を引き起こす内皮細胞由来弛緩因子として発見されました。その後、脳における一酸化窒素の役割が明らかになり、神経伝達、学習と記憶のプロセス、神経発生、神経変性疾患におけるガスとしての役割が判明しました。
一酸化窒素の合成
一酸化窒素は化学的にはNOと表記され、ドットは分子がフリーラジカルであり、高い反応性を持つことを表しています。一酸化窒素は、ガス状麻酔薬である亜酸化窒素(N2O)や、排気ガスに含まれる汚染物質である二酸化窒素(NO2)と混同されることがありますが、これらは哺乳類では内因的に合成されません。細胞内で一酸化窒素を生成する特定の酵素が存在し、それが一酸化窒素合成酵素です(図1.9-2)。この酵素は、アミノ酸のアルギニンから窒素を抽出し、酸素原子と反応させることで一酸化窒素を生成します。この酵素はニコチンアミドアデニンジヌクレオチドリン酸(NADPH)を利用し、副産物としてシトルリンを生成します。
一酸化窒素合成酵素には、それぞれ異なる場所と活性化パターンを持つ3つの異なる酵素型が存在します。神経型一酸化窒素合成酵素(nNOS)は、BredtとSnyderによって最初に発見された形態であり、脳の主要な形態です。神経型NOSはニューロン、特に皮質、海馬の歯状回、線条体、小脳のニューロンで発現しています。nNOS含有ニューロンは皮質ニューロンのわずか1%に過ぎませんが、その神経突起は非常に広範囲に分布しており、ほとんどすべてのニューロンがnNOS含有神経終末と接触しています。nNOS酵素活性は、補助タンパク質であるカルモジュリンを介したカルシウムレベルによって著しく増強されます。したがって、一酸化窒素は、カルシウムレベルが一時的に増加するニューロンの脱分極に続いて合成される可能性があります。
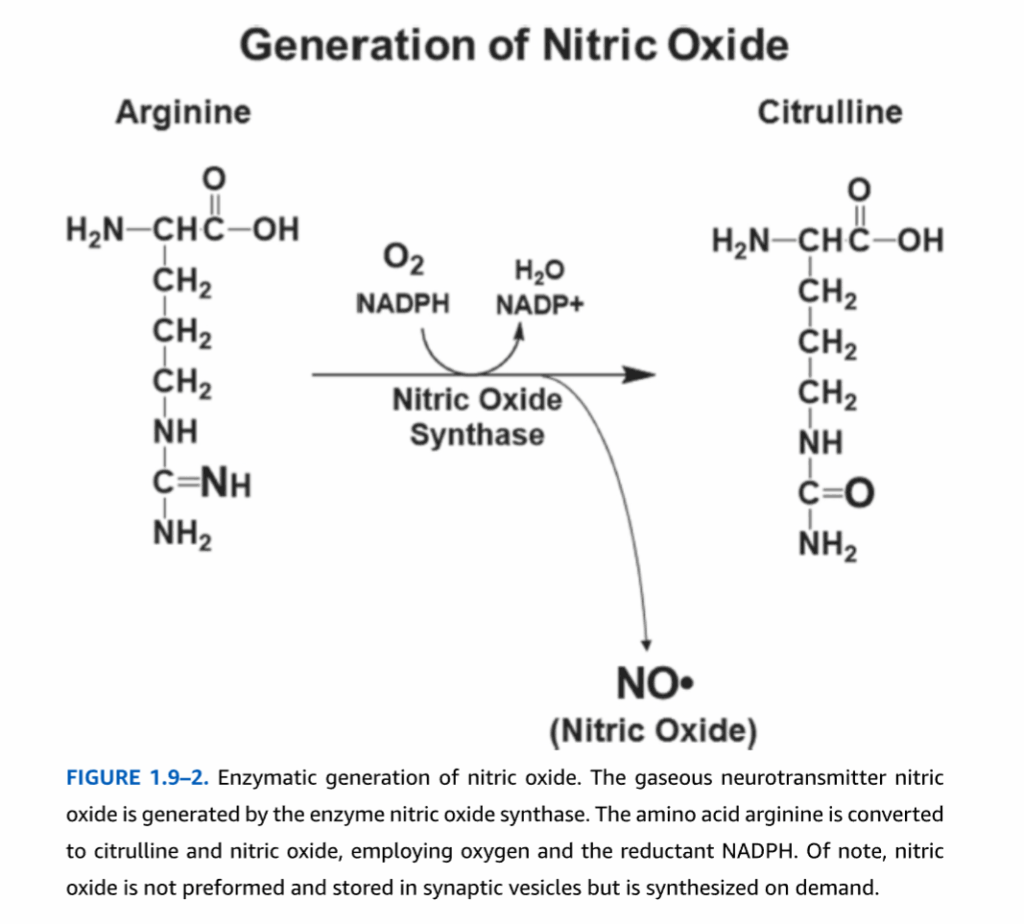
図1.9-2:一酸化窒素の酵素的生成
ガス状神経伝達物質である一酸化窒素は、一酸化窒素合成酵素によって生成されます。アミノ酸のアルギニンは、酸素と還元剤であるNADPHを用いて、シトルリンと一酸化窒素に変換されます。注目すべきは、一酸化窒素がシナプス小胞に前形成されて貯蔵されるのではなく、必要に応じて合成されるという点です。
内皮型NOS(eNOS)は主に血管に存在し、血管の弛緩と拡張を可能にする上で重要な役割を果たします。ニトログリセリンとニトロプルシドナトリウムは、一酸化窒素への変換を介して血管拡張作用を発揮します。eNOSの活性は、リン酸化と細胞内カルシウムの増加によって増強されます。3番目のNOSである誘導型NOS(iNOS)は、多くの組織にごく微量に存在します。しかし、そのレベルは、特に炎症などの非常に多様な細胞ストレスによって強く増加します。脳内では、主にグリア細胞で誘導されますが、ニューロンでも誘導されます。
一酸化窒素の作用機序:サイクリックGMP経路
学習や記憶などの脳機能の長期的な変化には、遺伝子やタンパク質の発現パターンの変化、樹状突起などの神経構造の物理的リモデリングを含む、非常に多様な細胞プロセスが関与しています。シグナル伝達は、神経の脱分極や受容体の活性化などの細胞外シグナルが、細胞機能の修飾につながるプロセスです。サイクリックグアノシン一リン酸(cGMP)は、膜受容体が活性化された後にグアニル酸シクラーゼによるその合成が刺激される、典型的な細胞内メッセンジャーです。cGMPはその後、生物学的効果を引き出すカスケードの一部としてタンパク質をリン酸化するプロテインキナーゼを活性化します。セロトニンなどの他の神経伝達物質の場合と同様に、一酸化窒素もニューロンでのcGMP産生を活性化します(図1.9-3)。典型的な神経伝達物質が膜受容体に結合したGタンパク質を介してグアニル酸シクラーゼを活性化するのに対し、一酸化窒素は、細胞質に存在する酵素型である可溶性グアニル酸シクラーゼを直接活性化します。グアニル酸シクラーゼの活性部位には、鉄原子が一酸化窒素によって結合されるヘムグループ補因子が含まれており、これによりタンパク質の立体構造変化とcGMPの産生が起こります。一酸化窒素は、ヘモグロビン/ミオグロビン、フェリチン、チトクロームP450など、他のタンパク質のヘムグループとも相互作用することができます。
一酸化窒素の作用機序:S-ニトロシル化経路
一酸化窒素が細胞に作用する2番目の方法は、S-ニトロシル化のプロセスです。このシグナル伝達メカニズムでは、一酸化窒素はタンパク質のシステイン残基の硫黄原子を直接修飾し、S-ニトロソチオール基を形成します(図1.9-4)。これにより、特定のタンパク質の活性が修飾されます。この修飾の生成には酵素は必要ありませんが、一部のタンパク質が細胞の異なる領域に修飾を運び、標的効果を引き出す目的のタンパク質に転移させるという証拠があります。ニトロシル化されたタンパク質は、この修飾に対する反応が様々で、活性化されるものもあれば、不活性化されるものもあります。
S-ニトロシル化のタンパク質標的の数は急速に拡大しており、シグナル伝達、プログラム細胞死、転写因子、細胞骨格タンパク質、イオンポンプ、イオンチャネルに関与する分子が含まれます。多くの場合、標的タンパク質中の単一のシステイン残基の修飾で、一酸化窒素がその活性を調節するのに十分です。S-ニトロシル化によって活性化される特定の標的には、L型カルシウムチャネル、カルシウム活性化カリウムチャネル、GABA-A受容体などがあります。ニトロシル化によって阻害されるタンパク質には、いくつかの種類のナトリウムチャネル、グルタミン酸神経伝達物質受容体のN-メチル-d-アスパラギン酸(NMDA)サブタイプ、およびいくつかの代謝酵素が含まれます。シグナル伝達の手段としてのS-ニトロシル化は、タンパク質のリン酸化とやや類似しています。どちらも細胞の活動を変化させるためにタンパク質機能を調節する可逆的な共有結合修飾だからです。S-ニトロシル化は、多くの脳タンパク質が神経型一酸化窒素の生成活動を介してニトロシル化されるため、記憶、学習、行動に関与する可能性があります。
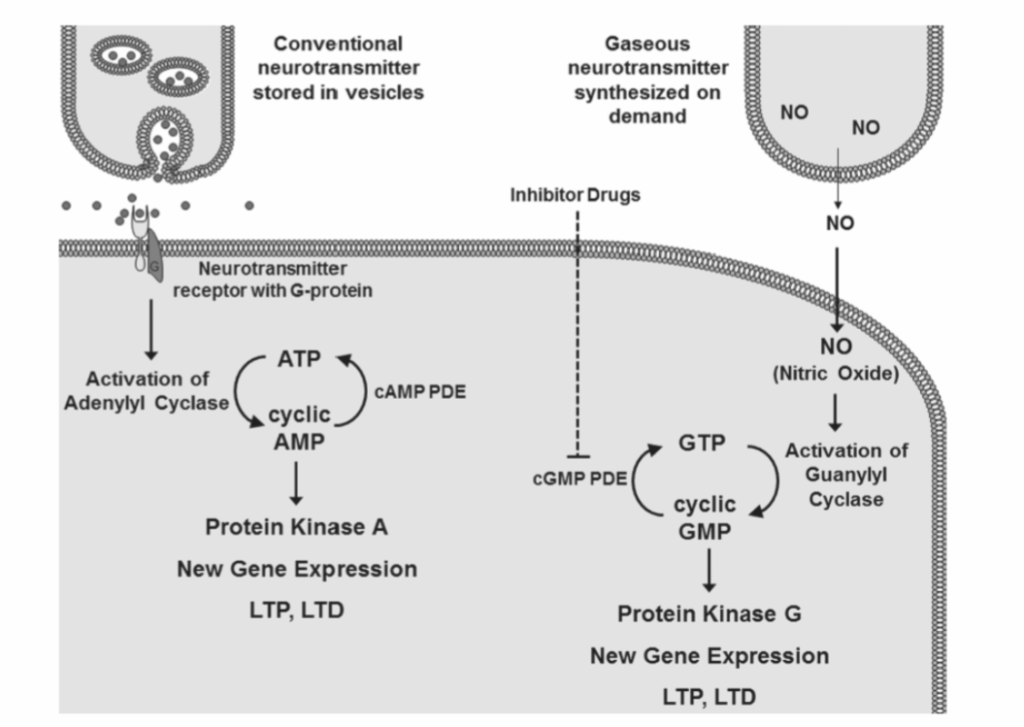
図1.9-3:サイクリックGMP産生を介した一酸化窒素(NO)の神経伝達物質およびシグナル伝達機能
ガス状の一酸化窒素は酵素的に生成され、隣接するニューロンに自由に拡散します(右上)。従来の神経伝達物質(左上)と比較して、NOはニューロンの表面膜上の特定の神経伝達物質受容体を介して作用しません。対照的に、NOはニューロン膜を自由に拡散し、グアニル酸シクラーゼという酵素を活性化します。この酵素はGTPをセカンドメッセンジャーであるサイクリックGMPに変換します。一酸化窒素の効果は、一部、サイクリックGMPによる神経プロテインキナーゼの活性化、新規遺伝子発現、および神経の**長期増強(LTP)と長期抑制(LTD)**への影響によって媒介されます。ATPはアデノシン三リン酸、PDEはホスホジエステラーゼです。
一酸化窒素と神経伝達
**長期増強(LTP)**は、シナプス前ニューロンの反復刺激がシナプス後ニューロンのより強力な発火につながるプロセスであり、学習と行動の変化の根底にあります。LTPの誘導はシナプス後NMDA受容体の活性化に依存しますが、LTPの維持はシナプス前メカニズムに依存します。NMDA受容体を介した神経伝達は、一部、一酸化窒素の活動を介してLTPを促進します。NMDA受容体の活性化は、細胞内カルシウムの増加につながり、シナプス後細胞における一酸化窒素合成とcGMP形成を促進します(図1.9-5)。
NOSの薬理学的阻害剤は、げっ歯類、鳥類、ミツバチモデルにおけるLTPの欠損を明らかにしており、一酸化窒素は短期記憶と長期記憶の両方の獲得に関与しているとされています。nNOSまたはeNOSのいずれかを欠損する遺伝子改変マウスは、海馬のLTPに変化を示しませんが、nNOSとeNOSの両方が欠損するマウスは、海馬のCA1領域でLTPの減少を示します。一方のNOSがもう一方の欠損を補償しているか、または両者がLTPで協調的に機能している可能性があります。腸管神経系の研究も、一酸化窒素が幽門括約筋の弛緩に果たす役割を明らかにしており、nNOS欠損マウスは幽門の著しい肥大を示します。一酸化窒素はまた、モノアミン作動性神経伝達を調節する可能性があります。ラットにおけるNOSの阻害はコカインとアンフェタミンの効果を増強しますが、一酸化窒素を増加させることによって逆の効果が観察されます。
一酸化窒素と行動
一酸化窒素神経伝達は行動に役割を果たす可能性があります。なぜなら、nNOS欠損雄マウスは攻撃的傾向が誇張され、性的活動が増加するからです。雌マウスでは、攻撃性が減少するため、逆のことが言えます。
末梢では、nNOSは陰茎の血管(海綿体を含む)を支配するニューロンに局在します。これらの神経の刺激は一酸化窒素を放出し、cGMPの形成、血管壁の弛緩と血管拡張、陰茎の充血、そして勃起の開始につながります。勃起の持続相も一酸化窒素に依存します。乱れた血流はeNOSのリン酸化と持続的な一酸化窒素産生につながります。勃起不全の治療に使用される薬物であるシルデナフィル(バイアグラ)、タダラフィル(シアリス)、バルデナフィル(レビトラ)は、陰茎のcGMPを分解する酵素である5型ホスホジエステラーゼを阻害することによって作用し(図1.9-3参照)、それによって一酸化窒素神経伝達と陰茎勃起を増強します。
図1.9-4:S-ニトロシル化を介した一酸化窒素(NO)シグナル伝達
NOによるグアニル酸シクラーゼの活性化に加えて、NOはS-ニトロシル化のプロセスを介してタンパク質機能を直接変化させることもあります。このプロセスは酵素的触媒作用を必要とせず、NOがタンパク質のシステイン残基の-SH基と反応し、その結果、-SNO修飾が生じ、タンパク質機能が変化します。一部のタンパク質はS-ニトロシル化後に強力な活性化を示しますが、他のタンパク質はこのプロセスによって阻害されます。
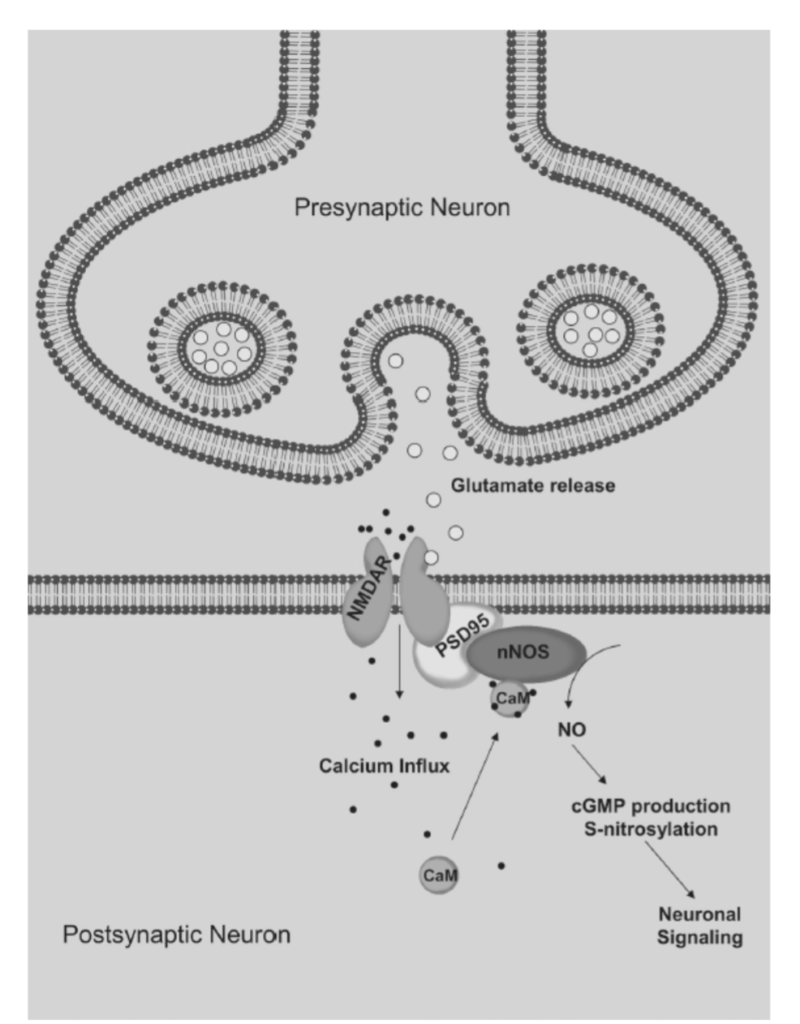
図1.9-5:NMDA受容体活性化後の一酸化窒素(NO)生成
シナプス前ニューロン(上)はグルタミン酸を放出し(図示せず)、NMDAグルタミン酸受容体を活性化してカルシウムがシナプス後ニューロンに入るのを可能にします。カルシウムはタンパク質である**カルモジュリン(CaM)に結合し、これが神経型一酸化窒素合成酵素(nNOS)**を活性化してNOを合成します。自由に拡散するガスであるNOは、サイクリックGMPの形成とS-ニトロシル化を介して標的ニューロンに作用します(図1.9-3および1.9-4)。
一酸化窒素が睡眠覚醒サイクルを調節する役割があることを示唆する多くの証拠があります。nNOSを発現するニューロンは、橋、背側縫線核、外側背側被蓋、脚橋被蓋など、レム睡眠を開始するいくつかの領域に存在します。動物モデルでは、一酸化窒素を放出する化合物の微量注入により、覚醒度が低下し、徐波睡眠が増加します。これと一致して、NOS阻害剤は徐波睡眠と急速眼球運動(REM)睡眠を減少させる傾向を示します。NOS欠損マウスの研究は、一酸化窒素が単に睡眠を促進する以上の複雑な役割を果たす可能性があることを示唆しています。nNOS欠損動物もREM睡眠の減少を示しますが、iNOS欠損マウスは逆の結果を示しており、NOS酵素アイソフォーム間の複雑な相互作用が示唆されます。
一酸化窒素と気分障害
NOSを発現するニューロンは、背側縫線核や前頭前野など、うつ病に関与する脳領域によく見られます。選択的セロトニン再取り込み阻害薬(SSRI)抗うつ薬が直接NOS活性を阻害できることから、一酸化窒素が抗うつ薬の反応に関与している可能性が示唆されています。さらに、強制水泳試験などの動物研究では、NOS阻害剤と可溶性グアニル酸シクラーゼ阻害剤が抗うつ薬様効果を達成できることが示されています。双極性障害患者では、健常対照者と比較して血漿中一酸化窒素レベルが上昇していました。しかし、うつ病患者では、一酸化窒素レベルの低下と一酸化窒素の副産物である血漿中亜硝酸塩の増加が報告されています。統合失調症とうつ病の患者の室傍核では、対照と比較してNOSの減少も報告されています。
成人脳で新しいニューロンが生成されるプロセスである神経新生は、気分障害の病態生理と抗うつ薬反応の両方に関与していることがますます認識されています。海馬の神経新生の増加は抗うつ薬反応と関連しており、海馬体積の減少は気分障害や不安障害のリスク因子となる可能性があります。セロトニン自体は海馬の神経新生を促進するようですが、一酸化窒素は神経新生を阻害することが判明しています。NOSの薬理学的阻害剤は、このプロセスの主要な部位である海馬の歯状回におけるセロトニンと神経新生の増加をもたらします。これらのNOS阻害剤はまた、歯状回におけるセロトニンの増加につながります。当然のことながら、nNOS欠損動物も歯状回における増殖の増加を示します。ステロイドがNOSの発現を誘導するようですので、一酸化窒素はこれらの薬剤で治療された人々にしばしば観察される気分や不安への影響に寄与する可能性があります。
一酸化窒素は、セロトニン、ノルエピネフリン、ドーパミンの神経終末における神経伝達を調節する能力について調査されています。一酸化窒素は、特定の脳領域とその活性化のタイミングに応じて、これらのニューロンの活動を増加させることも減少させることもできるようです。
一酸化窒素と統合失調症
一酸化窒素は、統合失調症の症状に寄与する候補分子として研究されてきました。遺伝学的研究により、nNOSと関連するタンパク質であるCAPONの統合失調症関連の一塩基多型(SNP)が特定されています。nNOSのSNPは、統合失調症と一貫して関連しているわけではありません。統合失調症患者の死後脳サンプルでNOSレベルの変化が報告されています。皮質、小脳、視床下部、脳幹で異常が指摘されていますが、特定の傾向は見られません。薬物未投与および薬物治療中の統合失調症患者の血小板でNOS活性の上昇が指摘されていますが、結果は一貫していません。一部の研究者は一酸化窒素活性の増加を、他の研究者は逆の結果を見出しています。剖検サンプルでは、統合失調症患者の前頭前野、海馬、側頭葉外側でNOSを発現するニューロンが異常な局在を示していることが判明しており、これは発生中のこれらのニューロン型の異常な移動と一致しています。ラットモデルでは、出生前のストレスが歯状回と海馬のNOS発現ニューロンの減少につながりました。
一酸化窒素の神経病理学的役割
一酸化窒素が様々な神経障害性事象に直接関与しているという豊富な証拠が存在します。細胞代謝の副産物であるスーパーオキシドは、一酸化窒素と反応してペルオキシナイトライト(化学式:ONOO-)を形成することがあります。この不安定で有毒な化合物は、タンパク質のチロシン残基(タンパク質ニトロ化と呼ばれるプロセス)やDNAと化学付加物を形成し、細胞機能不全につながります。虚血性脳卒中による細胞死は、一部、グルタミン酸NMDA受容体の過剰刺激(興奮毒性と呼ばれるプロセス)によって媒介されます。NMDA活性化によって産生される一酸化窒素は、この興奮毒性による神経細胞死の大部分を媒介しているようです。nNOSの遺伝子欠損を持つマウスでは、脳卒中による損傷が減少します。
S-ニトロシル化も脳内の病理学的プロセスに関与しています。パーキンソン病のパーキンタンパク質の変異は、早期発症型パーキンソン病と関連しています。パーキンはE3ユビキチンリガーゼであり、タンパク質にユビキチン分子を付加し、細胞プロテアソームでの破壊を標的とします。散発性パーキンソン病、つまり早期発症変異がない場合、一酸化窒素はパーキンタンパク質をニトロシル化し、その保護的なE3ユビキチンリガーゼ機能を阻害することができます。したがって、一酸化窒素シグナル伝達の過剰は、細胞機能に不可欠なタンパク質を妨害することにより、パーキンソン病におけるドーパミン作動性ニューロンの機能不全と細胞死を誘発する可能性があります。アルツハイマー病では、脳のタンパク質、脂質、炭水化物の過剰な酸化が古くから認識されていますが、過剰な一酸化窒素によるニトロ化ストレスも関与しているようです。**プロテインジスルフィドイソメラーゼ(PDI)**は、病気で発生するアミロイド線維などの誤って折りたたまれたタンパク質の蓄積と戦うのに役立つ細胞保護タンパク質です。アルツハイマー病とパーキンソン病の脳の両方で、PDIのS-ニトロシル化がその細胞保護機能を妨げています。
一酸化窒素が神経変性プロセスに関与していることが発見されたことで、完全な症状の発症前に一酸化窒素によって引き起こされる細胞成分の損傷を検出するなど、診断プロセスの改善の可能性が高まります。さらに、疾患の発症から保護する重要な神経タンパク質への損傷を軽減するよう設計された薬物が開発される可能性があります。しかし、一酸化窒素の合成を完全に、そして非特異的に阻害または刺激することは、全身に広範な活動を持つため、重大な副作用を引き起こす可能性が高いです。
一酸化窒素は、体内で最も集中的に研究されている化合物の一つであり、様々な臓器で多面的な活動を持っています。一酸化窒素は脳、血管系、免疫系において生理学的な役割を果たしていますが、病気にも関与する複雑で不完全に理解された分子です。しかし、一酸化窒素と同様に高い反応性を持つ酸素も、酸化損傷などの疾患病原性に関与する能力を持つ一方で、生命に不可欠な存在でもあります。
一酸化炭素
一酸化炭素(化学式:CO)は、燃焼反応に由来する大気汚染物質として最もよく知られていますが、ヒトから細菌に至るまで、様々な生物で生理学的に産生されています。かつては代謝反応の有毒な副産物と考えられていましたが、一酸化炭素は、脳やその他の臓器における様々な生理学的プロセスを調節することがますます認識されています。これらの多様な効果には、嗅覚神経伝達、血管弛緩、平滑筋細胞増殖、血小板凝集の調節などが含まれます。
一酸化炭素は、生理学的濃度での活動よりも、その毒性作用で遥かによく知られています。それはヘモグロビン内のヘム分子に強く結合し、カルボキシヘモグロビンを形成します。カルボキシヘモグロビンはもはや組織に酸素を輸送することができません。1日1〜2箱の喫煙者は、通常、ヘモグロビンの3%〜8%がカルボキシヘモグロビンであり、非喫煙者は2%未満です。急性一酸化炭素中毒の後、カルボキシヘモグロビンが5%〜10%の場合、注意と認知機能の障害が関連付けられ、30%〜50%のカルボキシヘモグロビンは、組織への酸素輸送の著しい低下につながります。
一酸化炭素の酵素的生成
一酸化炭素は、ヘムオキシゲナーゼ(HO)の作用によるヘムの代謝中に生成されます。この酵素は、酸素、NADPHの還元当量、および電子供与体であるチトクロームP450還元酵素を利用して、ヘムの炭素環を開き、一酸化炭素として1つの炭素断片を放出します(図1.9-6)。この反応では、緑色の色素であるビリベルジンと遊離鉄も生成されます。ビリベルジンは黄色の色素であるビリルビンに変換されますが、これは一酸化炭素と同様に、もはや単に有毒な副産物とは見なされていません。生理学的濃度では、ビリルビンは非常に強力な抗酸化物質であり、その前駆体であるビリベルジンに部分的にリサイクルされることがあります。
HOには3つの形態が存在します。HO1はiNOSと同様に、通常は非常に低いレベルで存在しますが、その発現は、酸化ストレス、炎症、ドーパミン、ステロイド、成長因子など、様々な刺激によって強力に誘導される可能性があります。実際、HO1は知られている中で最も容易に誘導されるタンパク質の1つです。HO2は誘導性のタンパク質ではなく、脳と精巣で主に発現しています。HO2は、皮質および海馬の錐体細胞、歯状回の顆粒細胞、嗅球、視床、視床下部、脳幹、小脳など、脳全体の離散した神経細胞集団で発現しています。HO3は、その重要性がよく理解されていないアイソフォームです。
一酸化炭素の分子作用
一酸化窒素と同様に、ガス状の一酸化炭素は膜を自由に拡散し、可溶性グアニル酸シクラーゼを直接活性化することができますが、その効力は一酸化窒素の約30分の1です。それに伴うcGMPの増加により、プロテインキナーゼが活性化され、細胞に対する一酸化炭素の多様な効果の一部が引き起こされます。HO2の発現はグアニル酸シクラーゼの発現パターンを密接に反映しており、両者が神経シグナル伝達の共通経路の一部であることを示唆しており、HO2の阻害剤はcGMPの生成を阻害します。nNOSの場合と同様に、HO2はカルシウム/カルモジュリンとリン酸化によって活性化され、神経の脱分極に応答して迅速に放出される神経伝達物質という重要な基準を満たします。一酸化炭素はまた、cGMPを必要としないメカニズムを介してp38 MAPキナーゼを活性化することができます。この重要なキナーゼは、炎症の抑制、細胞増殖、プログラム細胞死(アポトーシス)など、様々な細胞効果を促進します。
一酸化炭素と神経伝達
一酸化炭素は、嗅覚受容の神経伝達に関与しているようです。匂い物質は一酸化炭素の産生とその後のcGMP合成を導き、匂い刺激への長期的な適応を促進します。一酸化炭素は、まだ試験されていない様々な知覚および認知プロセスを調節する可能性を秘めています。同様に、ラットの網膜では、長時間の光曝露がHO1発現、一酸化炭素産生、およびcGMPシグナル伝達の増加につながります。一酸化炭素は慢性疼痛への適応にも関与する可能性があります。HO2欠損動物は、慢性疼痛刺激への曝露後、痛覚過敏およびアロディニアの減少を示します。したがって、一酸化炭素は疼痛知覚の閾値を調整する可能性がありますが、その効果が中枢神経系または末梢神経系(PNS)のいずれで生じるかは不明です。cGMP産生促進の役割とは別に、一酸化炭素はBKcaチャネルに直接結合して開くこともあります。
胃腸神経系では、一酸化炭素は非アドレナリン性非コリン作動性(NANC)神経刺激と**血管作動性腸管ペプチド(VIP)**に応答して、内肛門括約筋を弛緩させる神経伝達物質として機能します。HO2を遺伝的に欠損させたマウスは、nNOS欠損動物と同様に、NANC神経伝達が50%減少することを示します。HO2とnNOSの両方を欠損するように育種されたマウスでは、NANC神経伝達が完全に失われることから、ガス状神経伝達物質のみによって媒介される生理学的プロセスが確立されます。
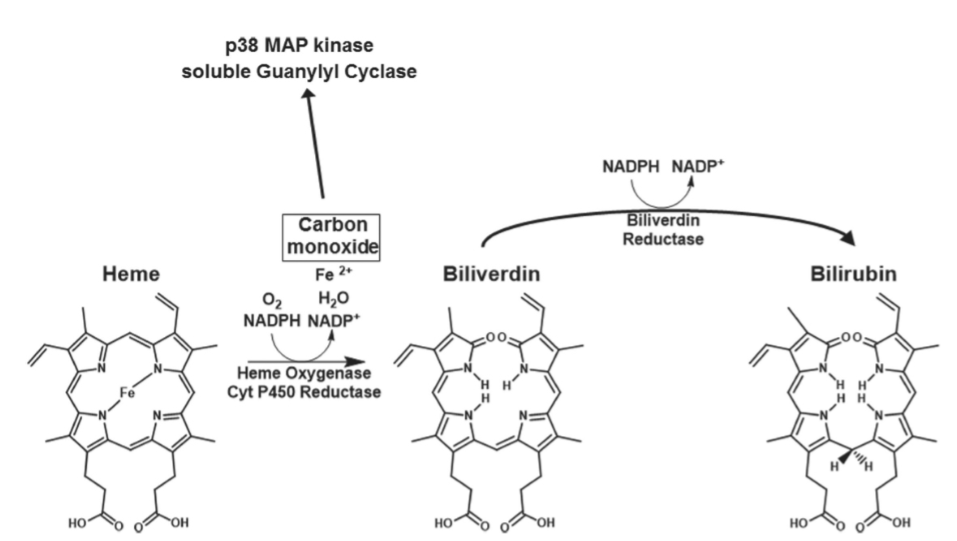
図1.9-6:非典型的神経伝達物質である一酸化炭素の合成
ガス状の一酸化炭素(CO)は、酵素であるヘムオキシゲナーゼを介してニューロンで酵素的に合成され、ヘムを分子ビリベルジンに変換し、遊離鉄(Fe)を放出します。一酸化窒素と同様に、COは神経小胞に貯蔵されず、神経膜を自由に拡散することができます。COも同様に可溶性グアニル酸シクラーゼを活性化し、p38 MAPキナーゼなどの複数の細胞内シグナル伝達分子の活性化につながります。COは、古典的なCO毒性が起こる濃度よりもはるかに低い濃度で、神経伝達物質およびシグナル伝達機能を発揮します。ニューロンにおけるこの経路の重要性は、2つの異なるヘムオキシゲナーゼ酵素が存在することによって強調されています。そのうちの1つは脳で主に発現しています。ビリベルジンは酵素ビリベルジン還元酵素を介してビリルビンに変換されます。COと同様に、ビリルビンももはや有毒な副産物の地位に追いやられず、重要な抗酸化物質である可能性があります。
一酸化炭素は、海馬のLTP(長期増強)の発達に関与しているとされていますが、証拠は矛盾しています。一酸化炭素と神経の強直性刺激は、興奮性シナプス後電位(EPSP)の増加につながります。一酸化炭素産生を阻害するHO阻害剤は、LTPの誘導障害とカルシウム依存性グルタミン酸神経伝達物質放出の減少につながります。しかし、HO2欠損動物はLTPに何らの違いも示しません。これらの異なる発見は、LTPにおけるHO1の役割、またはHO阻害剤がLTP誘導に重要な他のいくつかのプロセスを非特異的にブロックする能力によって説明されるかもしれません。
一酸化炭素のその他のシグナル伝達の役割
視床下部におけるHO2の発現により、研究者たちは一酸化炭素がペプチドホルモンの放出を調節できるかどうかを検証しました。動物モデルでは、一酸化炭素が視床下部からのオキシトシンとバソプレシンの両方の分泌を阻害することが判明しました。細胞培養システムでは、一酸化炭素が視床下部細胞からの副腎皮質刺激ホルモン放出因子(CRF)の放出を阻害することを示唆していますが、毒性に近いレベルでは逆の作用、すなわちCRF放出を刺激します。
ヘム代謝と一酸化炭素産生は、概日リズムで調節されているようで、睡眠覚醒サイクルの調節における役割と一致しています。哺乳類の転写因子NPAS2はBMAL1と結合して複合体(NPAS2/BMAL1)を形成し、これはClock/BMAL1と共に、periodおよびcryptochromeタンパク質の転写を活性化します。Periodとcryptochromeは二重の機能を持っており、概日リズム機構を阻害する一方で、NPAS2/BMAL1とClock/BMAL1もブロックして周期的な回路を形成します。NPAS2は一酸化炭素と結合できる2つのヘム部分を含んでおり、これによりNPAS2/BMAL1複合体がDNAに結合し、転写を調節する能力が阻害されます。
一酸化炭素は、毒性レベルでは、酸素よりも高い親和性でヘモグロビンに結合することで酸素輸送を阻害することがよく知られています。驚くべきことに、一酸化炭素自体は、頸動脈体が酸素を感知するメカニズムにおいて生理学的な役割を果たしています。頸動脈体のグロームス細胞で発現するHOは、一酸化炭素の生成に酸素を基質として使用します(図1.9-6参照)。酸素レベルが低下すると、一酸化炭素の産生も低下し、これにより頸動脈体が酸素を感知する閾値がリセットされます。この分子メカニズムは、一酸化炭素による頸動脈体BKイオンチャネルの調節を介して起こる可能性があります。
ヘムオキシゲナーゼ経路の神経保護的役割
HO2を遺伝的に欠損させたマウスは、外傷性脳損傷や脳卒中損傷に対する感受性が増加することを示します。HOの神経保護機能は、HOがアミロイド斑に存在するため、アルツハイマー病で損なわれている可能性があります。毒性のあるアミロイドβ断片の供給源である**アミロイド前駆体タンパク質(APP)**は、HOの神経保護機能に結合して阻害することができ、早期発症型アルツハイマー病と関連するAPP変異体は、HO機能をブロックする上で最も強力です。
硫化水素:最新のガス状メッセンジャー分子
一酸化炭素とビリルビンがその生理学的機能が認識されるまで毒性があるという評判であったように、同様の物語がガスである硫化水素についても展開されています。細菌や下水処理場からの悪臭を放つ有毒な排出物として依然として広く認識されていますが、硫化水素(化学式:H2S)は、重要な神経調節物質および非典型的神経伝達物質であることが今後証明されるかもしれません。
少なくとも2つの異なる酵素が硫化水素を生成できます。それは、シスタチオニンβ合成酵素(CBS)とシスタチオニンγリアーゼ(CSE)です。それぞれがシステインと水を硫化水素、ピルビン酸、アンモニアに変換する同じ反応を触媒します。興味深いことに、各酵素は独立した反応も触媒します。CBSはホモシステインとセリンをシスタチオニンに変換し、CSEはこれを使ってシステイン、アンモニア、2-オキソ酪酸を生成します。一酸化窒素と同様に、硫化水素も細動脈の拡張によって血圧を調節します。CSE欠損マウスは高血圧であり、硫化水素治療は血圧を低下させます。
脳内では、硫化水素は160マイクロモルもの高濃度で存在し、脳機能の調節における役割と一致しています。CBSは脳内に豊富に発現していますが、CSEは検出されません。他のガス状神経伝達物質を生成する酵素と同様に、CBSはカルシウム/カルモジュリンによって活性化されます。CBS欠損マウスは海馬のLTPが変化しています。硫化水素はまた、NMDA受容体電流を増強し、低レベルでは神経保護作用を持っています。
環境中の硫化水素は、用量依存的に様々な効果を引き起こします。ヒトは0.1〜1ppm(百万分率)で臭いを検出し、100〜150ppmでは嗅神経が麻痺します。320〜530ppmでは肺水腫が発生し、530〜1,000ppmでは強力な中枢神経系(CNS)活性化、呼吸停止、昏睡、死を引き起こします。極めて高いレベルは工業用化学合成や石油精製施設で発生し、より穏やかなレベルは下水処理施設、間欠泉、温泉、地熱サイトで見られます。
エンドカンナビノイド:マリファナから神経伝達へ
大麻、ヘンプ、ハシシ、マフェン、あるいは様々な俗語で知られているかどうかにかかわらず、マリファナは数千年にわたって人類によって栽培され、利用されてきました。そのリスクと利益が同等であるかどうかについては長年の議論がありますが、マリファナが脳にどのような影響を及ぼすかという謎の一部が解明されたのは最近数十年のことです。利用者が経験する「ハイ」、すなわち陶酔感と落ち着きは、ヒトの脳に内因性のカンナビノイド、つまりエンドカンナビノイドが関与する神経経路に大麻が作用することに関連しています。
大麻の最初の記述された薬用用途は、紀元前約2700年の中国の皇帝神農の薬局方にまで遡り、彼は様々な病気の治療にその使用を推奨していました。この頃には、有害な特性も明らかであり、大量のヘンプの実を摂取すると「悪魔が見える」ようになったり、利用者が「霊と交信し、体を軽くする」ようになったりするとされていました。何世紀にもわたって、大麻はインドで食欲増進剤として用いられ、習慣的なマリファナ利用者はいまだに「お腹が空く(the munchies)」という現象によく馴染んでいます。
長年にわたり、マリファナの活性成分であるカンナビノイドがその精神作用効果を発揮するメカニズムは謎に包まれていました。化学者たちは、植物油の多くの成分から大麻の精神活性成分を分離しようと努めました。1940年に最初に解明されたのはカンナビノールで、これは現在、他のカンナビノイドの酸化生成物であり、精神活性化合物ではないと認識されています(図1.9-7)。しかし、構造が手に入ると、化学者たちは精神活性特性を持つ合成カンナビノイドを合成できるようになりました。数年以内に、テトラヒドロカンナビノールが大麻の活性成分である可能性があることが明らかになりました。化学分離技術の進歩に続き、1964年にメコウラムとガオニは、大麻の精神作用のほぼすべてを担う化合物である**デルタ-9-テトラヒドロカンナビノール(THC)**を特定しました。THC酸は植物性THCの主要な形態であり、これは大麻を喫煙するときのように加熱すると容易にTHCに変換されます。
脳内エンドカンナビノイドシステムの発見
推定によると、マリファナのタバコ(「ジョイント」)を1本吸うと、20〜80マイクログラムのTHCが脳に到達します。これに対し、ヒトの脳全体には100〜200マイクログラムのノルエピネフリン神経伝達物質が存在することを考えてみてください。1960年代には、THCがその精神作用効果をどのように発揮するかについて、少なくとも2つの学説がありました。1つは、THCが吸入揮発性麻酔薬と同様の方法で作用するというもので、つまり、特定の受容体は存在せず、ニューロン膜に一般的な影響を及ぼすか、神経伝達物質受容体に広範な作用を及ぼすというものでした。競合する学説は、カンナビノイドに対する特定の受容体が脳内に存在すると仮説を立てましたが、これらの化学物質の親油性のために特定が困難でした。より水溶性の高い新規カンナビノイドが合成され、1980年代後半に、これによりCB1と名付けられた特定のカンナビノイド受容体の発見が可能になりました。
脳内にカンナビノイド受容体が存在することが知られるようになると、研究コミュニティは、そのような受容体が植物カンナビノイドの作用を可能にするためだけに進化するとは考えにくいと感じました。実際、1970年代にパートとスナイダーがオピオイド受容体を発見した後、これらがモルヒネ薬の目的のために進化したのではなく、その直後に発見された内因性エンケファリン神経伝達物質の標的として進化したことがすぐに明らかになりました。CB1受容体の内因性脳内リガンドの探索が進行中で、そのような物質の存在は内因性カンナビノイドであると仮説が立てられました。これは、1992年にメコウラムらが、脳内で内因的に産生され、カンナビノイド受容体を活性化し、神経伝達物質として機能する脂質であるアナンダミドの発見を報告した際に真実であることが証明されました(図1.9-8)。この物質の名前は、**サンスクリット語の「アナンダ(ananda)」**に由来し、「至福」と訳されます。アナンダミドはまた、自発運動の阻害、フリーズ(静止)発作の促進、痛覚過敏の軽減、体温の低下など、物質がヒトに精神活性特性を持つかどうかを一般的に予測する様々な動物行動試験においてTHCを模倣することができました。
その後まもなく、いくつかの追加のエンドカンナビノイド、2-アラキドノイルグリセロール(2-AG)、N-アラキドノイルドーパミン(NADA)、2-アラキドノイルグリセロールエーテル(ノラジンエーテル)、およびビロダミンが発見されました(図1.9-8参照)。いくつかの異なるエンドカンナビノイドが存在する理由は、カンナビノイド受容体であるCB1とCB2に対する親和性の違いにあるかもしれません。アナンダミドはCB1受容体に対する選択性が最も高く、次いでNADAとノラジンエーテルが続きます。対照的に、ビロダミンはCB2受容体を好み、CB1では部分作動薬活性しか持ちません。2-AGはCB1とCB2を区別しないようです。
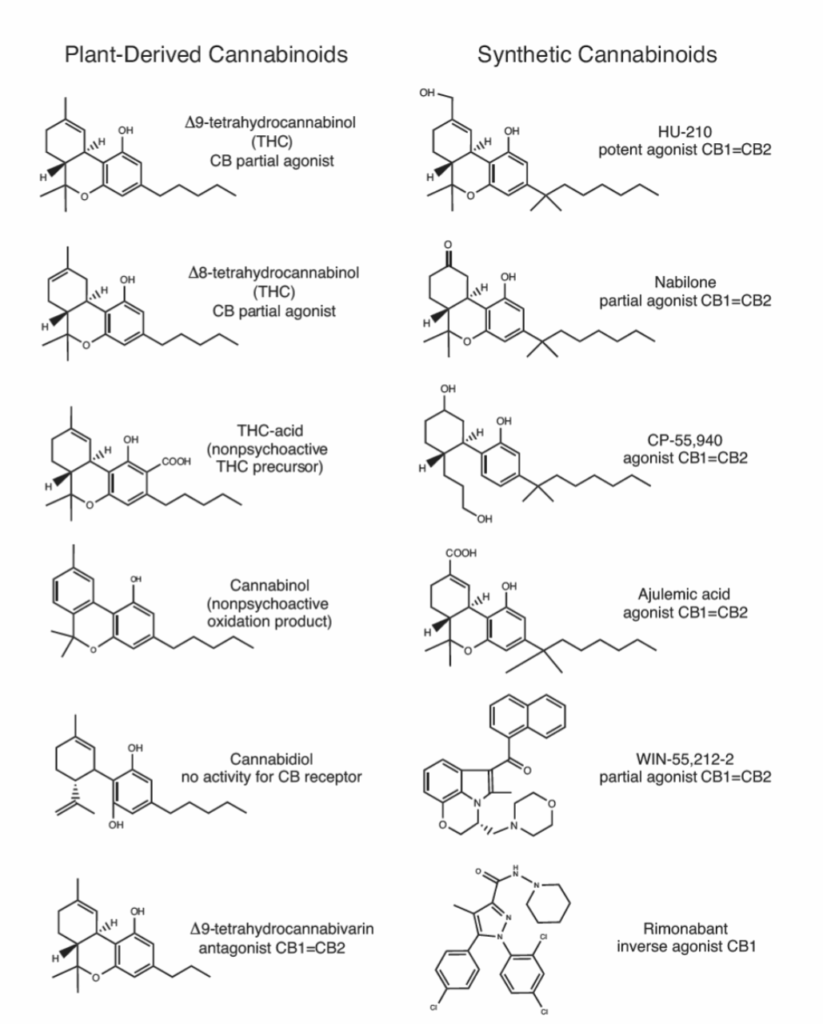
図1.9-7:植物由来および合成カンナビノイドの選択
THCは大麻の主要な精神活性成分です。薬物のリモンナバン(アコンプリア)は、カンナビノイドCB1受容体の強力な逆作動薬です。FAAHは脂肪酸アミドヒドロラーゼです。
エンドカンナビノイドの生合成
アラキドン酸は、エンドカンナビノイド、プロスタグランジン、ロイコトリエンの生合成のための構成要素として利用され、細胞膜や他の細胞内膜のリン脂質中に存在します。アナンダミドの合成には、2つの酵素の連続的な作用が必要です(図1.9-9)。最初の反応では、酵素NATがリン脂質からNAPEを生成するために、アラキドン酸側鎖をホスファチジルエタノールアミン(PE)に転移させます。2番目の反応では、酵素NAPD-PLDがNAPEをアナンダミドに変換します。NAPEはすでに哺乳類の膜の天然成分であるため、神経伝達にとって最も重要なのは、アナンダミドを生成する2番目のステップです。
2-AGの生合成にも2つの酵素が必要です。最初のステップでは、中間位置にアラキドン酸を含むリン脂質が、酵素ホスホリパーゼCの作用により**sn-1-アシル-2-アラキドノイルグリセロール(DAG)に変換されます。2番目の反応では、2つの特定のジアシルグリセロールリパーゼ(DAGL)**のいずれかを介して2-AGが生成されます。他のエンドカンナビノイドの生合成酵素は未定義です。
エンドカンナビノイドは、ガス状神経伝達物質の場合と同様に、後で使用するためにシナプス小胞に貯蔵されるのではなく、必要に応じて合成されます。シグナル伝達分子が神経伝達物質と見なされるための重要な基準は、神経の脱分極がその放出につながるべきであるという点です。脱分極は細胞内カルシウムの増加につながり、これがエンドカンナビノイドの合成と放出を促進します。このメカニズムは、NAPE-PLDおよびDAGLのカルシウム活性化によって部分的に説明され、それぞれアナンダミドと2-AGの生合成が増強されます。
ニューロンで生成されたエンドカンナビノイドは、カンナビノイド受容体に作用するためにシナプス間隙を横切る必要があります。THCと同様に、エンドカンナビノイドは非常に親油性であるため、脳脊髄液には溶けにくいです。エンドカンナビノイドがシナプス間隙を横切って標的ニューロン内に入ることを可能にする特定のエンドカンナビノイドトランスポーターが存在するという仮説が立てられています。
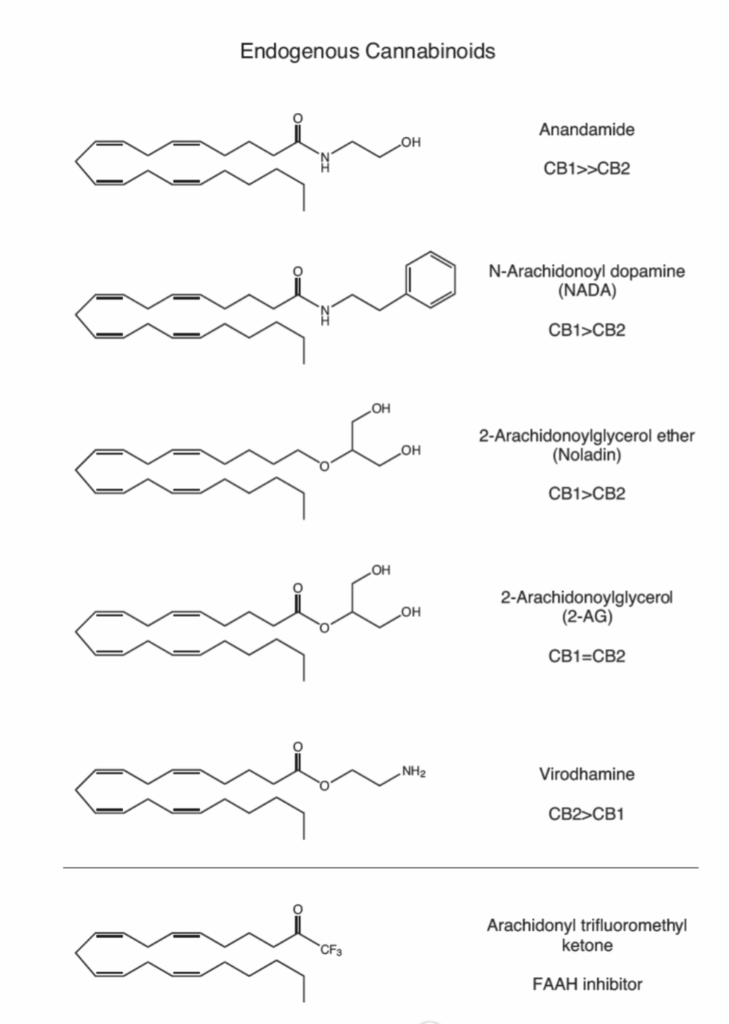
図1.9-8:内因性カンナビノイド
哺乳類の脳には少なくとも5種類のエンドカンナビノイドが存在し、それぞれがCB1およびCB2カンナビノイド受容体に対する親和性が異なります。これらはすべて、必須のオメガ-6脂肪酸であるアラキドン酸に由来しており、アラキドン酸はプロスタグランジンやロイコトリエンの生成における前駆体でもあります。
エンドカンナビノイドの不活性化
神経伝達物質は通常、放出するニューロンからの再取り込み、またはアセチルコリンがアセチルコリンエステラーゼによって加水分解されるように、高特異的な酵素による分解によって不活性化されます。エンドカンナビノイドの分解を標的とし、その神経伝達を減衰させる酵素は少なくとも2つ存在します。**脂肪酸アミドヒドロラーゼ(FAAH)は、アナンダミドをアラキドン酸とエタノールアミンに変換します(図1.9-9参照)。FAAHはCB1受容体が優勢な脳領域に存在し、アナンダミドが生成されるシナプス後ニューロンに局在します。アナンダミドの急速な分解は、THCと比較してその効力が比較的低い理由の一部を説明しています。FAAHがアナンダミドの不活性化に果たす役割を裏付けるように、FAAHを欠損するノックアウトマウスでは、アナンダミドが15倍に増加しますが、2-AGは増加しません。これらのマウスは、アナンダミドの分解が減少しているため、外因性アナンダミドに対する行動応答が大きくなります。エンドカンナビノイド2-AGはFAAHによって不活性化されますが、シナプス前ニューロンに存在するモノアシルグリセロールリパーゼ(MAGL)**によっても不活性化されます。
FAAHの薬理学的阻害剤は、動物モデルで鎮痛作用を持ち、不安を軽減しますが、不動、体温低下、食欲増進といったTHCの望ましくない効果はありません。このような薬理学的戦略は、**モノアミン酸化酵素阻害剤(MAOI)やカテコール-O-メチルトランスフェラーゼ阻害剤(COMTI)**に類似しています。うつ病で用いられるMAOIは、セロトニンや他のモノアミンの分解を阻害することでセロトニンを増加させ、COMTIはドーパミンや他のカテコールアミンの分解を阻害することで同様の役割を果たします。
カンナビノイド受容体
神経機能におけるその重要性を強調するように、CB1受容体は脳内で最も豊富なGタンパク質共役型受容体である可能性があります。これらは、大脳基底核、小脳、海馬、視床下部、前帯状皮質、大脳皮質、特に前頭前野で最高の密度で存在します。大量のTHCを投与されたヒトまたは動物は、自発運動の減少と奇妙で不自然な姿勢での凍結を伴うカタレプシーを発症します。大脳基底核と小脳におけるカンナビノイドの作用は、これらの行動と関連している可能性があり、統合失調症におけるカタトニー症状を理解する上で関連性があるかもしれません。
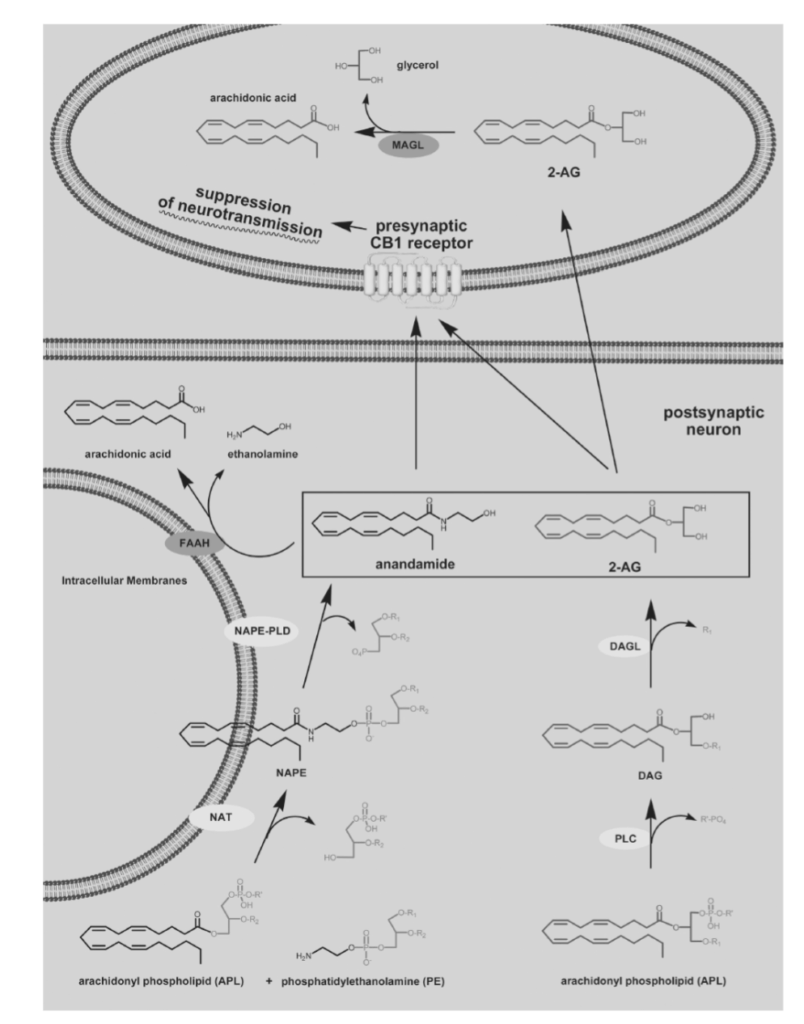
図1.9-9:エンドカンナビノイド(アナンダミドと2-アラキドノイルグリセロール(2-AG))の逆行性神経伝達
アナンダミドは、神経伝達のために二段階のプロセスで必要に応じて合成されます。酵素NATがリン脂質(APL)からアラキドン酸鎖をホスファチジルエタノールアミン(PE)に転移させ、NAPEを生成します。その後、2番目の酵素であるNAPE-PLDがアナンダミドを生成します。2-AGも同様に、酵素PLCとDAGLによって二段階で合成されます。シナプス後ニューロンで生成されたエンドカンナビノイドは、シナプスを横断してシナプス前CB1受容体を活性化し、シナプス前ニューロンの神経伝達を抑制します(ただし、場合によってはシナプス前ニューロンの活性化も起こります)。2-AGは主にシナプス前ニューロンでMAGLによって不活性化されますが、アナンダミドはシナプス後ニューロンでFAAHによって分解されます。
(図中の略語:PE, phosphatidylethanolamine; APL, arachidonyl phospholipids; NAT, N-acyltransferase; NAPE, N-arachidonoyl-phosphatidylethanolamine; NAPE-PLD, N-arachidonoyl-phosphatidylethanolamine phospholipase D; FAAH, fatty acid amide hydrolase; MAGL, monoacylglycerol lipase; PLC, phospholipase C; PDE, phosphodiesterase; DAG, diacylglycerol; DAGL, diacylglycerol lipase; R1-R3, various acyl or alkyl side chains of phospholipids; R’, side chain of phospholipid head group.)
カンナビノイド受容体
CB1受容体は主に軸索と神経終末に存在し、神経の樹状突起や細胞体にはほとんど見られません。これらの受容体は、シナプス間隙のシナプス後側よりもシナプス前側に局在する傾向があり、神経伝達の調節に関与していることを示唆しています。もう一つのカンナビノイド受容体であるCB2は、主に免疫系の白血球表面に発現していますが、少量が脳にも存在すると考えられています。
神経伝達への影響
カンナビノイドCB1受容体はGタンパク質と関連しており、その細胞内シグナル伝達は、一部、アデニル酸シクラーゼの阻害を介して媒介されます。これにより、重要なセカンドメッセンジャーであるサイクリックアデノシン一リン酸のレベルが低下します。CB1受容体の活性化は、カリウムチャネルの活性化とN型カルシウムチャネルの阻害にもつながります。カルシウムは神経伝達物質の放出に不可欠であるため、カンナビノイドはこのメカニズムを介して神経伝達をブロックすることができます。カンナビノイド受容体はまた、マイトジェン活性化プロテインキナーゼを活性化します。
細胞培養モデルや脳スライスを用いた研究では、カンナビノイドがGABA、ノルエピネフリン、アセチルコリンなど、様々な神経伝達物質の放出をブロックすることが示されています。ノルエピネフリンとアセチルコリンは興奮性神経伝達物質である傾向があり、カンナビノイドによるそれらの放出阻害は、全体として抑制効果を持つと予想されます。しかし、GABAは抑制性神経伝達物質であり、カンナビノイドによるその阻害は全体として興奮性効果につながるため、カンナビノイドは状況によって神経伝達に複雑な影響を与える可能性があることを示しています。カンナビノイドはまた、脳内エンドルフィン神経伝達物質の放出を増加させ、依存症や学習に関連する「報酬中枢」である側坐核におけるドーパミン放出を増加させるようです。エンドカンナビノイドは、**LTP(長期増強)やLTD(長期抑制)**を含む様々な形態のシナプス可塑性に関与しているとされています。
エンドカンナビノイドによって調節される逆行性伝達
シナプス後ニューロンがシナプス前ニューロンの活動を調節できることは、神経科学者にとって長年の間、明らかでした。例えば、シナプス前ニューロンによる神経伝達物質のさらなる放出を抑制することなどです。エンドカンナビノイドは、シナプス後ニューロンから拡散してシナプス前ニューロンに作用する逆行性メッセンジャーとして、現在までに最も有力な候補かもしれません。発生段階では、カンナビノイド合成に関与する酵素は、シナプス前ニューロンとシナプス後ニューロンの両方で発現していますが、成熟した脳では、エンドカンナビノイドの合成は主にシナプス後ニューロンで行われるようです。このことは、それらが逆方向に作用し、シナプス前ニューロンの神経伝達を調節する可能性があることを示唆しました。
ドーパミンやグルタミン酸を含む典型的なシナプス前ニューロンは神経伝達物質を放出し、シナプス後ニューロンの脱分極につながります。このシナプス後ニューロンはエンドカンナビノイドを放出し、それがシナプス間隙を横切って拡散し、シナプス前ニューロンからのさらなる神経伝達物質放出を抑制します。このようなメカニズムはげっ歯類の海馬で確認されており、CB1受容体拮抗薬はシナプス前ニューロンの抑制をブロックします。同様に、CB1受容体を遺伝的に欠損させたマウスは、シナプス前GABAニューロンの海馬抑制を失います(このプロセスは**脱分極誘発性抑制抑制(DSI)としても知られています)。逆もまた証明されており、エンドカンナビノイド破壊阻害剤は海馬における逆行性神経伝達を増強します。エンドカンナビノイドは、LTDや脱分極誘発性興奮抑制(DSE)**など、他の神経伝達プロセスにも関与しています。エンドカンナビノイドが神経伝達物質放出を抑制する能力は、神経伝達が調節される重要な一般的メカニズムである可能性があります。
不安と気分におけるエンドカンナビノイド
エンドカンナビノイド神経伝達は、不安の重要な調節因子である可能性があり、大麻使用者はTHCの鎮静効果を頻繁に述べます。エンドカンナビノイドシステムのシグナル伝達の喪失は、動物研究で不安様状態を促進するようです。CB1受容体欠損動物は、ストレスや新しい環境に曝された際に、より顕著な不安行動を示します。
マルシカーノらは、カンナビノイドシグナル伝達が苦痛な記憶を忘れるのに役割を果たすことを示唆しました。音と電気ショックを組み合わせたマウスモデルでは、CB1受容体を欠損する動物は、通常見られる消去(ショックが伴わない音単独への反応で凍結しなくなること)を示しませんでした。したがって、エンドカンナビノイド神経伝達は、苦痛な記憶に関連する不安を「忘れる」能力を媒介する可能性があります。CB1拮抗薬も同様の効果を示しました。扁桃体は多くの不安応答に関与しており、エンドカンナビノイドはこの脳領域に作用して不安を軽減する可能性があります。これを裏付けるように、マウスが音に曝された直後に、扁桃体でアナンダミドと2-AGのレベルが増加することが判明しました。
FAAHノックアウトマウスはエンドカンナビノイドを分解する酵素を欠いており、アナンダミドレベルの増加と行動試験における不安の減少の両方を示します。エンドカンナビノイドレベルの増強は、不安に対する治療標的となる可能性があります。FAAH阻害剤はアナンダミドの分解を減少させ、動物の不安様行動を軽減します。「強制水泳試験」や「尾懸垂試験」はマウスにおけるうつ病の完璧なモデルではありませんが、FAAH阻害剤は動物がこれらのストレスに対処する能力を改善し、これは抗うつ薬治療でも観察される利点です。
エンドカンナビノイド経路は、心的外傷後ストレス反応や恐怖症の理解における魅力的な標的となる可能性があります。ヒト被験者の脳内エンドカンナビノイドレベルを日常的に測定することはまだできませんが、このモデルは、体重減少戦略として期待されるカンナビノイド受容体遮断薬であるリモンナバンの臨床試験によって裏付けられています。リモンナバンは不安とうつ病の増加という副作用が頻繁にあり、2007年のメタアナリシスでは、リモンナバンを投与された患者が、うつ病や不安のために治療を中止するリスクが著しく高かったと報告されています。これらの精神科的副作用は、既往歴のある患者を除外した研究であったにもかかわらず発生しており、このシステムが不安と気分の調節において重要な役割を果たしていることを強調しています。
大麻の使用は気分への鎮静効果と関連していますが、一部の利用者では逆説的な不安を経験するため、エンドカンナビノイドは気分障害に役割を果たす可能性があります。自殺で死亡したうつ病患者の死後研究では、前頭前野でCB1受容体が増加していることが判明しており、これはアルコール依存症に関連する自殺の研究でも観察されています。遺伝子関連研究では、CB1受容体と精神疾患の関連が模索されており、結果は混在していますが、パーキンソン病におけるうつ病や、アルコール依存症患者における注意欠陥障害との関連が示唆されています。
FAAH阻害剤は、ヒト試験でまだ基準を満たしていません。SSR-411298はうつ病の治療において不十分な改善しか示さず、PF-04457845は膝関節炎の痛みを改善できませんでした。BIA 10-2474の第1相臨床試験では、90人の参加者のうち5人が出血性壊死性脳病変を経験し、1人が死亡しました。これは、この経路へのヒトの薬理学的介入の難しさを示しています。
依存症
エンドカンナビノイドシステムは、依存症の理解における魅力的な標的となる可能性があります。CB1受容体を欠損するマウスは、当然ながらカンナビノイドの行動効果に対して耐性を示します。しかし、彼らはオピオイドへの依存と離脱も減少するように見えます。カンナビノイドがドーパミン放出を増加させることで、オピオイドシステムとカンナビノイドシステムの間でさらなる相互作用も発見されています。側坐核は依存症に関与する主要な報酬領域であり、このドーパミン放出はμ-オピオイド受容体を必要とするようです。これらの受容体を薬理学的に阻害すると、カンナビノイドがドーパミン放出を増加させる能力がブロックされます。アルコールを好むラットはFAAH活性が低下しており、これはより大きなカンナビノイドシグナル伝達を示唆しています。CB1受容体拮抗薬はアルコール摂取量を減衰させますが、FAAHを阻害するとアルコール摂取量が増加します。さらに、CB1欠損動物もアルコール摂取量が減少するように見えます。ヒトのFAAHにおける単一アミノ酸変異が薬物乱用と関連していることが判明しており、この異常な酵素は野生型よりも不安定であるようです。
精神病におけるエンドカンナビノイド
大麻を大量に使用すると、精神疾患の既往がない個人でも精神病症状を引き起こす可能性がありますが、これが薬物のみによるものなのか、精神病に対する潜在的な脆弱性によるものなのかは不明です。大麻の使用はしばしば統合失調症の精神病を悪化させ、大量使用は統合失調症の発症と関連しています。ただし、この関連は、最終的に統合失調症を発症する人々の症状の加速された発達であると示唆する人もいます。それにもかかわらず、カンナビノイドシグナル伝達がドーパミン放出を増加させるように見えるため、エンドカンナビノイドシステムは統合失調症の病態生理学に影響を与えます。ドーパミンD2受容体の拮抗薬として作用する薬は、しばらくの間、統合失調症治療の構成要素として残る可能性が高いでしょう。
統合失調症患者の脳脊髄液でアナンダミドレベルの上昇が認められており、薬物未投与の患者の追跡調査でも同様の報告があります。統合失調症患者の血液でもアナンダミドレベルの上昇が認められており、そのような上昇は臨床的改善とともに正常化しました。
統合失調症患者の死後脳サンプル、特に背外側前頭前野と帯状皮質でCB1受容体レベルの上昇が報告されています。CB1受容体の多型が統合失調症と関連していることを特定した研究者もいますが、関連性を見出せなかった研究者もいます。さらに、そのような多型がCB1機能に与える影響は不明です。
摂食
薬物摂取後、THC使用者は食欲増進(「マンチー」)を経験し、大麻は何世紀にもわたって食欲増進剤として利用されてきました。この効果は、視床下部に存在するCB1受容体に依存する可能性があります。動物が食物を奪われると、視床下部と辺縁系でエンドカンナビノイドレベルが増加します。CB1受容体を遺伝的に欠損させたマウスは、高脂肪食を与えられても肥満になりにくくなります。同様に、CB1受容体拮抗薬であるリモンナバンは、カンナビノイドシグナル伝達をブロックすることで体重減少を促進するようです。3,000人以上の肥満患者を対象とした臨床試験では、1日20mgのリモンナバンで治療された患者は1年後に6.3kgの体重減少を示しましたが、プラセボ群では1.6kgでした。吐き気が最も一般的な副作用として報告されました。2007年の臨床試験のメタアナリシスでは、リモンナバン治療で全体として4.7kgの体重減少が報告され、体重減少薬のオルリスタット(2.9kg)やシブトラミン(4.2kg)を上回りました。残念ながら、不安やうつ病などの重篤な副作用がその臨床使用を妨げました。
脳損傷と疼痛への影響
外傷性脳損傷のマウスモデルにおいて、2-AGは神経保護作用を示し、脳浮腫、梗塞サイズ、細胞死を軽減し、機能的転帰を改善します。アナンダミドも多発性硬化症のモデルで脳損傷から保護し、ヒト患者ではアナンダミドの産生が増加しています。カンナビノイド作動薬HU-211の研究では、頭部外傷後の臨床的改善がより迅速になりました。FAAH阻害剤はパーキンソン病のマウスモデルで運動症状を改善し、これはカンナビノイドがドーパミン神経伝達を増加させることによるものと考えられます。
エンドカンナビノイド経路を介した神経伝達は、疼痛知覚の調節においてますます重要性が認識されています。THCとカンナビノイド作動薬は、熱傷から神経損傷、炎症に至るまで、急性および慢性疼痛の動物モデルで有効であることが証明されています。CB1受容体はこれらの効果に重要な役割を果たしており、CB1拮抗薬であるリモンナバンが投与されると、カンナビノイド薬の鎮痛効果は失われます。同様に、CB1受容体を遺伝的に欠損するマウスではTHCの鎮痛効果が失われます。
ストレスは、負傷した軍人が顕著な痛覚耐性を示す現象(ストレス誘発性鎮痛として知られる)のように、疼痛知覚の低下と長らく関連付けられてきました。エンドカンナビノイドシステムがこれらの効果を媒介する可能性があります。動物モデルでは、ストレス後のアナンダミドと2-AGの産生が明らかになり、これらの動物ではストレス誘発性鎮痛がCB1遮断薬リモンナバンによってブロックされます。多発性硬化症患者600人以上を対象としたプラセボ対照無作為化研究では、ザジチェクらが経口THC投与が運動能力と疼痛の改善につながることを発見しました。しかし、THCは子宮摘出後の術後疼痛にはほとんど効果がありませんでした。
疼痛知覚のエンドカンナビノイド調節は内因性オピエートシステムとは異なると考えられますが、両経路は重複する神経経路を共有している可能性があります。このことは、CB1遮断薬リモンナバンとオピエート受容体をブロックするナロキソンを用いた研究で示されています。リモンナバンはTHCやカンナビノイドによる鎮痛を減衰させますが、モルヒネへの反応を部分的にしかブロックしません。しかし、オピエートでは逆であり、ナロキソンはモルヒネ誘発性鎮痛をブロックするだけでなく、THCやカンナビノイド薬の鎮痛も部分的にブロックします。カンナビノイドとオピエート薬の組み合わせは、動物モデルで相乗的な鎮痛効果を示します。
カンナビノイドが鎮痛効果を中枢神経系を介して発揮すると当初考えられていましたが、動物モデルでは、カンナビノイドの局所投与も有効であることが示されており、中枢神経系での発現が最小限であるCB2受容体に選択的な薬物も含まれます。
エンドカンナビノイドは、CB1およびCB2受容体が関与しないメカニズムによっても痛覚感受性に影響を与える可能性があります。アナンダミドとNADAの両方は、知覚神経に存在するバニロイド受容体(TRPV-1としても知られる)として知られるカルシウムチャネルも活性化することができます。この同じ受容体は、唐辛子の辛味を引き起こすカプサイシンによって活性化されることで注目されます。したがって、エンドカンナビノイドは、CB1およびCB2受容体を介して鎮痛を促進する一方で、TRPチャネルを介して痛みを増加させるという相反する機能を果たす可能性があります。CB2受容体は主に末梢で発現していますが、死後分析ではアルツハイマー病患者の脳でその上方制御が明らかになっています。
新規カンナビノイド薬の急速な開発により、THCの典型的な効果をすべて引き出すのではなく、特定の症状を標的とすることが可能になるかもしれません。例えば、アジュレミック酸は鎮痛作用と抗炎症作用を示しますが、精神作用の副作用が限定的であるという利点があります。この化合物の無作為化臨床試験では、アジュレミック酸は慢性神経因性疼痛の軽減に有効であることが証明されました。
末梢への影響
カンナビノイドは、局所的なCB1受容体を介して血管平滑筋の直接的な弛緩を引き起こします。この血管拡張は眼の結膜にも及び、一部の大麻利用者では「充血した目」の状態になります。カンナビノイドによる眼動脈の弛緩は、高眼圧の状態である緑内障の治療に役立つ可能性があり、腎臓のCB1受容体の活性化は腎血流を改善することができます。全身の血圧調節における役割は証明されておらず、リモンナバンで治療された人やCB1受容体を欠損する動物では血圧は変化しません。カンナビノイドシグナル伝達は異所性妊娠にも関連している可能性があり、CB1欠損マウスは卵管に多くの胚を保持します。
合成カンナビノイド
1980年代以降、何百もの化学的に異なる合成カンナビノイドが生成されてきました(図1.9-7参照)。当初は研究目的で生成されましたが、利用者が陶酔効果を求めるために人気が高まりました。政府はしばしばそのような使用を迅速に禁止してきましたが、そのたびに新しい異なる化学物質が出現しました。これらやその他多くの物質は、「スパイス」や「K2」と呼ばれるレクリエーションドラッグに様々に混ぜられ、通常は喫煙または経口摂取され、THCの標準的な薬物検査では検出されません。**デルタ-8-テトラヒドロカンナビノール(デルタ-8-THC)**はTHCの密接な構造類似体です。大麻植物には少量しか存在しませんが、化学者によってカンナビジオールを前駆体として一般的に作られています。その精神作用効果はTHCよりも弱く、これは一部にはTHCよりも約6倍効率が低いCB1受容体への結合に関連しています。**ドロナビノール(マリノール)**は、化学療法誘発性の悪心と嘔吐を治療するためにFDAの承認を受けた合成THCです。ナビロンはCB1とCB2の部分作動薬であり、かつて同様の用途で承認されましたが、その後市場から撤退しました。
非精神作用性カンナビノイド
THCが大麻の主要な精神作用成分であるにもかかわらず、多くの非精神作用性カンナビノイドも興味深い特性を持ち、神経伝達を調節する可能性があります。カンナビジオールは潜在的な治療効果を提供し、TRP-V1受容体を刺激し、エンドカンナビノイドの分解に影響を与えるようです。カンナビジオールはまた、炎症性関節炎のマウスモデルで保護効果を示し、抗けいれん作用があるとされています。結果は混在していますが、精製されたカンナビジオールも抗精神病作用を発揮する可能性があります。ただし、植物大麻の使用による正味の効果は、THC成分のために統合失調症の症状を悪化させることが一般的です。テトラヒドロカンナビバリンはCB1受容体の拮抗薬である植物カンナビノイドです。これは、患者が植物由来の大麻を使用しているのか、テトラヒドロカンナビバリンを含まない処方THCを使用しているのかを区別するための候補マーカーです。
