
1.10 神経栄養因子
JIHYE KIM, M.D., FRANCIS S. LEE, M.D., PH.D., AND MOSES V. CHAO, PH.D.
ニューロトロフィンは、神経細胞および非神経細胞の増殖、分化、生存、および死に影響を与えるユニークなポリペプチド成長因子ファミリーです。ニューロトロフィンの効果は、その利用可能なレベル、膜貫通型受容体への結合親和性、および受容体活性化後に刺激される下流シグナル伝達カスケードに依存します。ニューロトロフィンの生物学的役割は当初、神経系の発達中に特徴づけられましたが、現在では、シナプス結合、シナプス構造、神経伝達物質放出、長期増強(LTP)、機械感覚、および痛みなどの成人神経系における複数の役割を持つことが明らかになっています。ニューロトロフィンレベルの変化は、アルツハイマー病(AD)などの神経変性疾患だけでなく、不安障害、うつ病、物質乱用などの精神障害にも関連付けられています。これらの成長因子の精神障害における役割の概念化において、抗うつおよび抗不安治療反応の媒介における主要な役割へと大きな転換が起こりました。これらの最近の知見は、精神障害の病因と治療において彼らが果たす役割について情報を提供しています。
ニューロトロフィンファミリー
多くのポリペプチド因子が、神経系の生存、成長、および分化に影響を与えます。神経成長因子(NGF)、脳由来神経栄養因子(BDNF)、ニューロトロフィン-3(NT-3)、およびニューロトロフィン-4(NT-4)から構成されるニューロトロフィンは、神経系で最もよく理解され、最も広く発現しています。ニューロトロフィンは、最初に前駆体またはプロニューロトロフィンとして合成され、成熟した活性タンパク質を放出するために切断されます。成熟タンパク質は、約12〜14 kDaのサイズで、安定した非共有結合ダイマーを形成し、発達中に非常に低いレベルで通常発現されます。プロニューロトロフィンの切断とプロセシングは、フューリンによるゴルジ装置、プロコンベルターゼによる濃密な小胞、およびプラスミンまたは選択的マトリックスメタロプロテイナーゼによる細胞外など、複数の部位で起こる可能性があります。
NGFは最初に特定された神経栄養因子であり、その分布は限定的です。末梢神経系(PNS)では、侵害受容および温度感覚に関与する交感神経ニューロンおよび感覚ニューロンに作用します。中枢神経系(CNS)では、NGFは前脳基底部のコリン作動性ニューロンの生存と機能促進します。これらのニューロンは海馬に投射し、ADで特に影響を受ける記憶プロセスにとって重要であると考えられています。
他のニューロトロフィンは、CNSでより広く発現しています。BDNFとNT-3は皮質および海馬構造で高く発現しており、複数のニューロン集団の生存と機能に関連付けられています。BDNFは、ストレス、損傷、神経刺激、および運動後に、はるかに高い濃度で存在します。ニューロンの生存と分化に加えて、BDNFは活動依存性可塑性に非常に反応します(「ニューロトロフィンとシナプス可塑性」のサブセクションを参照)。他のニューロトロフィンの場合と同様に、あらゆる痛み、損傷、または炎症は、NGF、BDNF、および他の炎症誘発性ポリペプチドの転写の有意な増加をもたらす可能性があります。
ニューロトロフィン受容体
ニューロトロフィンは、2つの異なる受容体の作用を介して細胞効果を発揮するという点でユニークです。それは、**トロポミオシン関連キナーゼ(Trk)**という受容体チロシンキナーゼ受容体と、**p75神経栄養因子受容体(p75NTR)**という腫瘍壊死因子(TNF)受容体スーパーファミリーメンバーです。Trk受容体は、細胞外リガンド結合領域、単一の膜貫通ドメイン、および高度に保存された細胞内チロシンキナーゼドメインから構成されます。p75NTRは、細胞外リガンド結合領域、単一の膜貫通ドメイン、およびデスドメインと呼ばれるタンパク質結合領域を含む細胞内部分から構成されます。
ニューロトロフィンはダイマーとしてTrkファミリーメンバーに結合し、受容体の二量体化と触媒チロシンプロテインキナーゼドメインの活性化を導きます。二量体化したTrk受容体は、いくつかの主要な細胞内チロシン残基を自己リン酸化し、これにより細胞内シグナル伝達カスケードが迅速に開始されます。これは、受容体上のリン酸化されたチロシンが、Srcホモロジー領域2(SH2)などのホスホチロシン結合モチーフを含む特定のアダプタータンパク質の結合のための認識部位として機能することで達成されます。特に、Shcアダプタータンパク質は、活性化されたTrk受容体を、ニューロトロフィンのほとんどの生物学的効果を媒介する2つの別々の細胞内シグナル伝達経路に連結します。
主要な生存経路はShcを介し、Trk受容体活性化をホスファチジルイノシトール-3-キナーゼ活性の増加に連結します。これは、次にもう1つのプロテインキナーゼであるAkt(プロテインキナーゼB)を活性化し、Aktは細胞のアポトーシス経路に複数の影響を与えます。また、Trk受容体活性化によるShcのリン酸化は、Rasおよびマイトジェン活性化プロテインキナーゼ(MAPK)活性の増加につながります。これらの事象は、サイクリックアデノシン一リン酸(cAMP)応答エレメント結合タンパク質(CREB)転写因子および哺乳類ラパマイシン標的複合体(mTORC)の誘導など、転写および翻訳の事象に影響を与えます。CREBは、細胞周期、神経突起伸長、およびシナプス可塑性に多数の効果をもたらします。mTORCは、局所的な活動依存性翻訳を促進することで学習と記憶を調節することが示されており、これによりLTPに伴うスパインのリモデリングにつながります。これらのメカニズムは、ケタミンを含む様々な薬物の抗うつ作用に関与しているとされています(関連サブセクション「気分障害の神経栄養仮説:ニューロトロフィンの治療可能性」を参照)。加えて、ホスホリパーゼ-C-γ(PLC-γ)は活性化されたTrk受容体に結合し、イノシトールリン酸の細胞内シグナル伝達カスケード放出とプロテインキナーゼCの活性化を開始します。Trk受容体活性化は、多数の下流シグナル伝達イベントにつながり、転写および翻訳プログラムの変化を引き起こします。
NGFはTrkAに最も特異的に結合し、BDNFとNT-4はTrkBに、NT-3はTrkC受容体に結合します(図1.10-1)。p75NTRは各ニューロトロフィンに結合できますが、Trkのその同種リガンドに対する親和性を調節する追加の能力を持っています。Trkとp75NTRは、それぞれ高親和性受容体と低親和性受容体として言及されてきました。しかし、TrkAとTrkBは成熟ニューロトロフィンに10$^{-9}から10^{-10}Mの親和性で結合するため、これは高親和性部位(Ka=10^{-11}$M)よりも低いため、これは正しくありません。また、NGFの前駆体型はp75NTRに高親和性結合を示します。NGFの低濃度に対するTrkを介した応答性は、p75NTRとTrkA受容体の相対レベルと、高親和性部位を形成するそれらの複合能力に依存します。受容体の比率が応答性、そして最終的には神経細胞数を決定できるため、これは重要です。
p75NTRとTrk受容体は直接互いに結合しませんが、2つの受容体の間に複合体が形成されるという証拠があります。これらの相互作用の結果、p75NTRによってTrk受容体にリガンド選択性が付与される可能性があります。特異性を生み出す1つの方法は、Trk受容体に対するリガンドのより大きな識別力を与えることです。例えば、BDNF、NT-3、およびNT-4/5はそれぞれTrkB受容体に結合できますが、p75NTRの存在下では、BDNFのみが機能的応答を提供します。同様に、NGFとNT-3の両方がTrkAに結合できますが、p75NTRはTrkAのシグナル伝達をNGFに限定し、NT-3には限定しません。したがって、p75NTRとTrk受容体は相互作用して、異なるニューロトロフィン間のより大きな識別力を提供します。
Trk受容体を介して作用するニューロトロフィンは、細胞の生存と分化を促進します。逆説的に、それらは細胞死を誘発することもできます。p75NTRは、発達中の細胞死および神経系の損傷後にアポトーシス促進性受容体として機能します。p75NTR発現の増加は、自然発生的な神経細胞死の期間中の胚性網膜および交感神経ニューロンにおけるアポトーシスの原因です。BDNFが交感神経ニューロンのp75NTRに結合すると急速な細胞死を引き起こしますが、NGFが同じニューロンのTrkA受容体に結合すると生存シグナルを提供します。ニューロトロフィンプロセシングの文脈では、プロニューロトロフィンは、p75依存性アポトーシスを誘発するにおいて、成熟NGFよりも効果的です。これらの結果は、ニューロトロフィンの生物学的作用がプロテアーゼ的切断によって調節される可能性があり、プロ型がp75NTRを選択的に活性化してアポトーシスを媒介し、成熟型がTrk受容体を選択的に活性化して生存を促進することを示唆しています。
神経栄養因子と発達
ニューロトロフィンは、その最初の発見以来、初期の発生段階における末梢神経系(PNS)で高く発現していることが観察されてきました。それらは、異なる発達段階における特定のニューロン集団の生存に不可欠です。これらの発見は、発達における神経栄養仮説の概念化につながり、これはPNSの発達における神経栄養因子の役割に対する機能的な説明を提供します。発達中、皮膚や筋肉などの同じ最終標的に近づくニューロンは、限られた量の標的由来神経栄養因子を巡って競合します。このようにして、神経系は最も競争力があり適切な接続のみを維持するように自身を形成します。標的細胞によって産生される限られた量の神経栄養分子を巡るニューロン間の競争が、選択的な細胞生存を説明します。この仮説からは2つの予測が生まれます。第一に、神経細胞の生存の有効性は、発達中に産生される栄養因子の量に依存するということです。第二に、応答性細胞集団における特定の受容体発現が、神経細胞の応答性を決定するということです。
ある意味で、ニューロトロフィンは神経栄養仮説によく適合します。なぜなら、多くの末梢神経細胞サブタイプが、自然発生的な細胞死の期間中に特定のニューロトロフィンに依存しているからです。さらに、発達中のPNSにおけるニューロトロフィンの生物学的効果の特異性は、ニューロトロフィン-リガンド-受容体複合体の細胞内位置によって調節され得ます。発達中、ニューロトロフィンは標的組織から産生され、放出されます。それらは小胞に取り込まれ、細胞体へ輸送されます。興味深いことに、ニューロトロフィンの生物学的効果は、神経終末から細胞体まで長距離にわたってシグナルが伝達されることを必要とします。したがって、神経栄養仮説の中心的なテーマは、神経細胞の生存と分化が、標的組織で産生される神経栄養因子の逆行性シグナル伝達に依存するということです。
PNSとは対照的に、発達中のCNSにおけるニューロトロフィンの役割は著しく異なり、これはおそらくTrk受容体の優勢な発現とp75NTRの限定的な発現によるものです。CNSでは、複数のTrk神経栄養因子受容体とその同種リガンドの重複する発現が、成人期まで十分に及ぶより多様な接続性を可能にします。注目すべきは、PNSとは異なり、CNSで発現する主要なニューロトロフィンはBDNFとNT-3であるということです。遺伝子ノックアウト研究から、それらの主要な機能は神経細胞の生存ではなく、神経細胞の分化とシナプス可塑性であることが明らかになっています。現在までに、CNSにおけるニューロトロフィン機能の多くの証拠基盤はBDNFに焦点を当てています。
可塑性におけるニューロトロフィンの役割を示す最初の例の1つは、視覚系の臨界期に関連する可塑性の調節に関して特定されました。視覚系におけるシナプス可塑性の発達調節は、皮質第4層における眼優位柱の形成によって示されます。このプロセスはBDNFによって強く影響され、抑制性γ-アミノ酪酸産生(GABA)介在ニューロンの成熟を調節し、それによって視覚可塑性の臨界期を調節します。BDNFを過剰発現するマウスは、視力の発達が加速され、臨界期の早期終了が見られることが示されています。また、視覚系可塑性に対するこれらの効果は、ニューロトロフィンに対する抗体やニューロトロフィン拮抗薬(例:ニューロトロフィンに結合するTrkB-IgG融合タンパク質)によってブロックされ、内因性ニューロトロフィンレベルの変化が臨界期の形成に劇的な結果をもたらすことを示しています。
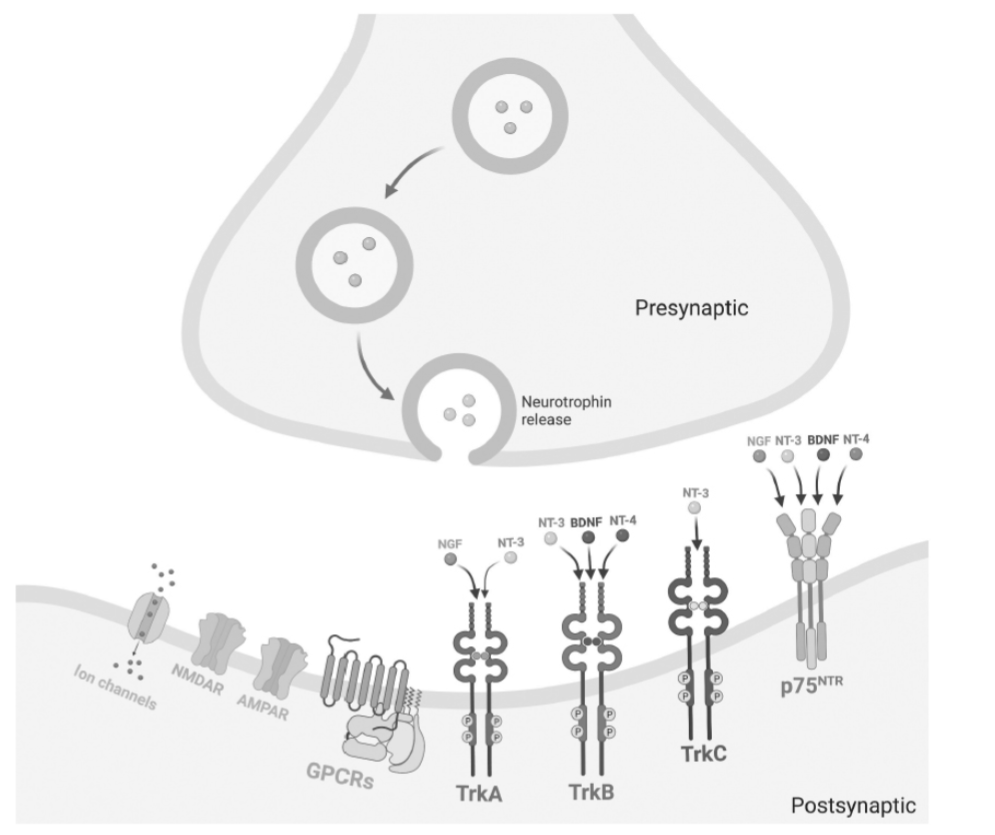
図1.10-1:シナプスにおけるニューロトロフィン
ニューロトロフィンはシナプス前側で放出され、特定のトロポミオシン関連キナーゼ(Trk)受容体に選択的に結合します。すべてのニューロトロフィンは**p75神経栄養因子受容体(p75NTR)**に結合します。**ニューロトロフィン-3(NT-3)とニューロトロフィン-4(NT-4)**は複数のTrk受容体に結合します。**神経成長因子(NGF)**はTrkAに結合し、**脳由来神経栄養因子(BDNF)**はTrkBに特異的に結合します。矢印の太さは、特定のニューロトロフィンと特定の受容体の結合親和性の強さを示します。(BioRender.comで作成)
BDNFは、恐怖制御の臨界期の調節にも関与していることが示されています。ヒトBDNF遺伝子の単一ヌクレオチド多型(rs62565)は、BDNFプロドメイン領域の66位でのバリン(Val)からメチオニン(Met)への置換を引き起こし、マウスにおいてBDNF分泌の減少と思春期周辺の前頭前野-海馬神経回路の発達障害につながることが示されています。思春期周辺のこの重要な期間における適切な発達の欠如は、成人期における条件付けられた恐怖反応を消去する能力の低下につながります。ヒトのBDNF MetアレルキャリアとヒトBDNF Metアレルを持つマウスの両方が、急性消去相において条件付けられた嫌悪刺激に対する反応の長期化を示し、これは初期の条件付けられた恐怖消去学習プロセスが効率的でないことを示唆しています。したがって、BDNF Metキャリアは、条件付けられた嫌悪刺激を消去するためにより集中的に調整された精神医学的治療(例えば、より長くまたはより集中的な暴露療法)から利益を得る可能性が高いでしょう。関連して、Metアレルを保有するヒトとBDNF Metマウスの両方は、様々な環境ストレス下に置かれた際に不安表現型が増加することを示します。この文脈において、BDNF Metアレルと人生のストレス要因および小児期の逆境との間の遺伝子-環境相互作用を調べた入手可能なすべてのヒトBDNF Metアレル研究の最近のメタアナリシスは、Metアレルがストレスの多い人生の出来事と小児期の逆境とうつ病との関係を有意に調節したことを示しました。要するに、BDNF Metアレルは、完全な反応を得るために個別化された精神医学的治療を必要とする可能性のある、うつ病および不安に基づく症状に対する遺伝的脆弱性を付与する可能性があります。
ニューロトロフィンとシナプス可塑性
BDNFの成人脳におけるシナプス可塑性の調節における役割は、海馬、前頭前野、線条体などの様々な脳領域で確立されています。多くの研究が、BDNF-TrkBシグナル伝達が成人脳のシナプス可塑性において重要な役割を果たすことを示しています。外因性BDNFの適用、遺伝子操作、または薬理学的阻害剤の遮断に基づいて、BDNF-TrkBシグナル伝達は、海馬における高周波刺激誘発性LTPの誘導を促進するが、必須ではないことが確立されています。ニューロトロフィンはまた、化学的LTPを誘発することも示されています。外因性BDNFの短時間適用は、海馬で長期的なシナプス増強を引き起こします。興味深いことに、BDNFの放出とBDNF依存性のスパインの再成長およびシナプス可塑性が学習と記憶に必要であるという強力な証拠があるにもかかわらず、この化学的「BDNF-LTP」は部分的にしか理解されていません。現在BDNF-LTPについて理解されていることは、細胞内カルシウムシグナル伝達と、細胞外シグナル調節キナーゼ(ERK)を含むいくつかの主要な下流経路が関与しているということです。したがって、出生後のCNSでは、BDNFはシナプス増強と神経伝達を調節するだけでなく、軸索と樹状突起の成長と分化も促進することができます。逆に、BDNFの前駆体であるproBDNFは、p75NTRの活性化により、海馬スライスにおけるLTDを促進することが示されています。外因性proBDNFの適用は、神経筋シナプスでシナプス抑制を誘発し、proBDNFが成熟BDNFのシナプス可塑性に対する作用と拮抗することを示唆しています。場合によっては、発達中の神経筋シナプスのように、proBDNFと成熟BDNFは、不活性な終末と活性な終末に対してそれぞれ潜在的な「罰」と「報酬」シグナルとして作用することが示されています。具体的には、活動依存性の細胞外プロテアーゼの活性化は、活性な終末におけるproBDNFから成熟BDNFへの変換につながり、シナプスの安定化をもたらします。逆に、p75NTRを発現する終末はproBDNFに結合し、「罰」シグナルを与えられて収縮します。proBDNFと成熟BDNFのこれらの相殺する効果は、p75NTRレベルが最も高い発達の初期段階で最も顕著であると考えられます。また、慢性ストレスとうつ病のげっ歯類モデルにおいて、proBDNFレベルが増加し、フルオキセチン治療で正常化することが示されています。しかし、大うつ病性障害(MDD)やその他の関連障害の患者における末梢proBDNFと成熟BDNFのレベルを比較したヒト研究は、概ねまちまちで結論が出ていません。したがって、患者のproBDNFと成熟BDNFのレベルが、精神疾患や治療反応の信頼できるバイオマーカーとして機能する可能性は低いでしょう。
ニューロトロフィンと代謝
BDNFとTrkBは、満腹感とエネルギーバランス調節の中枢制御において重要な役割を果たすことも確立されています。両方のプロセスは、主に視床下部領域におけるCNS回路の組み合わせ、および中枢および末梢組織によって産生されるホルモンや神経伝達物質によって調節されます。BDNFまたはTrkBのいずれかの発現が減少した機能喪失型遺伝子ノックアウトマウスの解析は、摂食量の増加と体重増加という一貫した表現型を示しました。最近の研究では、BDNFとTrkBを発現する別々の神経細胞集団を含む核の異種クラスターである**室傍視床下部(PVH)**が、食欲の神経栄養因子依存性抑制の主要な脳領域として特定されています。前部PVHにおけるBDNFの選択的欠失は、有意な過食と体重増加につながり、対照動物のPVHへの組換えBDNFの注入は摂食量を抑制します。別の一連のPVHニューロンにおけるTrkBの欠失も、有意な過食と体重増加につながり、これはTrkBを発現するこれらのニューロンの神経活動を低下させることによって模倣できました。
BDNF-TrkBは、基礎代謝、身体活動、体温調節を含むエネルギー消費の調節にも関与しています。同じ前部PVHサブ領域におけるBDNFの欠失は、エネルギー消費と運動活動の低下につながりました。これは、これらのニューロンから熱産生を制御する脊髄交感神経節ネットワークへの投射を介している可能性が高いです。逆に、対照動物のPVHへの組換えBDNFの注入は、エネルギー消費の増加につながりました。BDNF-TrkBは他の脳回路でシナプス可塑性の増強を媒介することが示されているため、BDNFによる食欲抑制とエネルギーバランスの調節は、これらの視床下部回路内のシナプス可塑性を調節している可能性が高いです。
並行して、ヒトの遺伝子研究では、BDNFおよびTrkBの突然変異と肥満および体格指数(BMI)の増加が直接関連付けられています。TrkBのキナーゼドメインにおける稀な新規ミスセンス変異、およびBDNFハプロ不全を引き起こす新規突然変異は、過食と重度の肥満の被験者で特定されています。肥満の測定値に関するゲノムワイド関連研究では、BDNF Val66Metアレルを含む、ゲノムワイドな有意性の基準を満たすBDNF遺伝子内またはその近くのいくつかのSNPが特定されています。最後に、抗精神病薬を服用しているヒトBDNF Metアレルキャリアの無作為化対照研究では、BDNF Metアレルが抗精神病薬関連肥満およびインスリン抵抗性の危険因子であることが示されました。これらのげっ歯類およびヒトにおける知見は、エネルギー消費と摂食行動の中枢制御におけるBDNFとTrkBの新規な役割を特定しています。これらのプロセスに関与する脳回路は、学習と記憶および気分調節における確立された役割とは異なりますが、シナプス伝達と可塑性の根底にある促進は、この形態の代謝調節に関与するそれらの異なる役割に共通するテーマである可能性が高いです。
神経変性疾患との相関
ニューロトロフィンは、様々な神経変性疾患および神経損傷疾患の病態生理に関与しているとされています。脳の急性および慢性の環境変化に対する反応は、神経機能の変化につながります。これらの臨床的相関の根底にある仮説、および神経栄養因子を用いた治療戦略の開発は、これらの疾患状態が以下の結果をもたらすと仮定しています。すなわち、罹患ニューロンに対するニューロトロフィンの利用可能性の低下、罹患ニューロン上のニューロトロフィン受容体の減少、および/または神経細胞生存率の低下です。これらの欠陥は、神経栄養因子の追加によって改善される可能性があります。
ニューロトロフィンの利用可能性の低下が神経精神医学的臨床問題の原因となる例として、**ハンチントン病(HD)が挙げられます。これは、線条体における中型有棘ニューロンの進行性の死を特徴とします。変異型ハンチンチンタンパク質の病原性効果を説明する一つのメカニズムは、皮質から線条体への順行性輸送の低下によって引き起こされるBDNFレベルの低下によるものです。同様の論理は、最も一般的な神経認知障害の一つであるアルツハイマー病(AD)**にも当てはまります。内嗅皮質や海馬のような脳の脆弱な領域にNGFまたはBDNFを追加すると、空間学習と記憶保持が増加します。これらの疾患状態では、外因性の神経栄養因子が、これらの神経系疾患の核心的な病態生理を治癒するのではなく、疾患状態に対する対症療法を提供すると仮定されてきました。よく定義された回路におけるニューロトロフィンとその受容体のより完全な細胞発現プロファイルは、異なる起源の変性疾患を標的とする戦略を提供するでしょう。
精神疾患との相関
神経栄養因子がシナプス結合、シナプス可塑性、および神経伝達に与える深遠な影響は、不安障害、うつ病、物質乱用などの精神疾患との関連性の基礎を形成してきました。ニューロトロフィンは、精神疾患の病態生理に関与する魅力的な分子中間体となりました。ニューロトロフィンへの環境的入力は、おそらく神経回路の変化、ひいては行動の変化につながる可能性があります。ニューロトロフィンが成人ニューロンの機能における転写プログラムを調節することで、長期的な変化を生み出すことができることが明らかになっています。これは、多くの精神医学的治療における治療作用の長い遅延を説明できるかもしれません。ここでも、臨床的相関は、疾患状態の表現型に寄与する神経栄養因子へのアクセスまたは応答性の欠陥が存在すると仮定しています。
大うつ病性障害
ニューロトロフィンの役割に関する最も強力な証拠は、うつ病の病態生理、特にストレスに関連するものから得られています。うつ病の場合、海馬などの脳領域におけるシナプス可塑性および神経細胞生存の根本的な調節不全があると考えられています。うつ病におけるニューロトロフィンの役割を示唆するいくつかの証拠があります。第一に、動物モデルでは、拘束ストレスが海馬におけるBDNFの発現を減少させます。加えて、慢性的な身体的または心理社会的ストレスは、特にげっ歯類および霊長類におけるCA3領域の海馬ニューロンの萎縮と死につながります。また、磁気共鳴画像法(MRI)研究では、うつ病または心的外傷後ストレス障害(PTSD)の患者が海馬体積のわずかな減少を示すことが示されています。しかし、これらのニューロンの萎縮および/または死がBDNFの利用可能性の低下に直接関連しているかどうかは不明です。さらに、すべての形態のうつ病がストレスと関連しているわけではありません。しかし、もし構造的リモデリングとシナプス可塑性がうつ病の細胞病態生理に関与している場合、BDNFはこれらの変化を媒介する魅力的な候補分子です。
第二に、海馬に外因的に投与されたBDNFは、2つの動物モデル(強制水泳および学習性無力感パラダイム)で、薬理学的抗うつ薬による慢性治療に匹敵する抗うつ効果を示しました。加えて、BDNFはin vitroおよびin vivoでセロトニン作動性ニューロンおよびノルアドレナリン作動性ニューロンに対して栄養効果を持つことも示されています。BDNFレベルが減少した変異マウスは、セロトニン作動性ニューロン機能の選択的低下と、セロトニン作動性異常と一致する対応する行動機能不全を示すことが示されています。
第三に、セロトニンおよびノルアドレナリン再取り込み阻害薬の抗うつ薬は、治療効果(10〜20日)と対応する時間経過で、BDNFとcAMP依存性転写因子であるCREBを上方制御します。CREB転写因子は、ニューロンにおけるBDNF遺伝子発現の誘導に関与しています。このcAMP経路への影響は、モノアミン抗うつ薬とニューロトロフィン間のつながりを提供します。抗うつ薬治療はまた、海馬におけるTrkB受容体の発現の増加をもたらし、これもこれらの治療の治療作用の延長された時間経過と並行しています。
特に、長期間のセロトニンおよびノルアドレナリン再取り込み阻害薬治療の効果は神経栄養因子シグナル伝達の強化を伴うにもかかわらず、中脳辺縁系ドーパミン系に外因的に投与されたBDNFは逆の効果、すなわちうつ病様の行動の増加を示すようです。加えて、このドーパミン回路におけるBDNFの除去は、社会的敗北パラダイムにおいて抗うつ効果を持つようです。これらの発見は、うつ病に関連する行動の側面を媒介するBDNFの役割の複雑さを強調しています。これらの研究は、うつ病治療の潜在的な治療標的として神経栄養因子システムをさらに調べるための枠組みを提供します。
気分障害の神経栄養仮説:ニューロトロフィンの治療可能性
最近の臨床試験は、神経変性疾患および精神疾患に対する神経栄養因子を使用する治療戦略を設計する上での限界を示しています。第一に、標的ニューロンに十分な量を物理的に送達することが主要な障害であることが明らかになりました。血液脳関門を容易に通過して神経栄養因子受容体を活性化するか、神経栄養因子の作用を増強する低分子化合物の開発は、初期段階にあります。第二に、ニューロトロフィンは神経活動に複数の影響を与えるため、神経栄養因子をCNSに無差別に「溢れさせる」ことは、てんかん活動などの望ましくない副作用につながる可能性があります。臨床試験では、BDNFの無規制な適用後にTrkB受容体の下方制御があることが指摘されており、これは最小限の治療効果に寄与している可能性があります。新しい戦略として、ウイルスベクターまたは遺伝子操作された前駆細胞の定位脳内注射を介して、より局所的かつ規制された神経栄養因子の適用が含まれる研究が進められています。このアプローチは現在、ADに適用されており、ADでは前脳基底部のコリン作動性ニューロンという明確な神経細胞集団が変性し、特定の神経栄養因子(NGF)に依存しています。
他の受容体シグナル伝達システムを介した神経栄養因子システムの活性化は、代替戦略を提供します。例えば、モノアミンGタンパク質共役受容体(GPCR)を介して作用する抗うつ薬は、ニューロトロフィンとニューロトロフィン受容体の両方の発現増加につながる可能性があります。重要なのは、モノアミンGPCRを発現するニューロンのみが、神経栄養因子またはTrk受容体の産生を増強するということです。他のGPCR、プリンアデノシン2A(A2A)受容体、および下垂体アデニル酸シクラーゼ活性化ペプチド(PACAP)神経ペプチド受容体が、in vitroで海馬ニューロンにおいてニューロトロフィンが存在しない状況でTrk神経栄養因子受容体をトランス活性化できることも示されています。したがって、低分子化合物はニューロトロフィンが存在しない状況でTrk受容体を活性化することができます。これらの結果は、低分子化合物が、特定のGPCRとTrk受容体を発現するニューロンを選択的に標的とすることにより、神経変性疾患の治療のための神経栄養効果を引き出すために使用できる可能性を示唆しています。
治療抵抗性うつ病および晩期うつ病に対する別のゴールドスタンダード治療法で、BDNFに依存することが示されているのは**電気けいれん療法(ECT)**です。げっ歯類では、長期的なECTがBDNF転写を増加させ、海馬ニューロンの出芽を誘発することが示されています。この効果は、BDNFレベルが低い変異マウスでは減弱しました。有望なげっ歯類研究にもかかわらず、臨床研究は、ECTがBDNFレベルを増加させる効果に関してより入り混じった結果を示しています。一部の研究ではECT治療で血清および脳脊髄液のBDNFレベルの増加が見られる一方で、他の研究ではそうではありません。治療反応のバイオマーカーとしてBDNFを調べようと試みた研究は、BDNFレベルがECT治療結果の予測因子としては不十分であると結論付けています。これはおそらく、BDNFが広範な遺伝的、生物学的、および環境的要因によって影響を受けるためです。ECTの臨床的に有意な治療効果がBDNF依存性であるかどうかを判断するには、より厳密に力を入れた研究が必要です。
ニューロトロフィンレベルを増加させる他の強力な方法には、特定の種類の抗うつ薬の使用が含まれます。様々なセロトニンおよびセロトニン-ノルエピネフリン再取り込み阻害薬の抗うつ薬は、数週間の投与後にのみ臨床的に有効です。新しいアプローチでは、速効性の非競合的N-メチル-D-アスパラギン酸(NMDA)受容体拮抗薬であるケタミンの効果が検討されています。ケタミンは、BDNF依存性メカニズムを介して急速な抗うつ反応を媒介することが示されています。ケタミンによるNMDA受容体の遮断は、真核生物伸長因子-2キナーゼ(eEF2キナーゼ)の不活性化につながります。これにより、BDNF翻訳のブロックが解除され、迅速なBDNFタンパク質合成(30分以内)が起こります。このBDNFの急速な上方制御は、その後、スパインのリモデリング、シナプス形成、およびシナプス可塑性の誘導を引き起こします。あるいは、ケタミンによるNMDA受容体の遮断は、急速で一時的なグルタミン酸バーストにつながり、それがAMPA受容体を刺激します。AMPA受容体の刺激は、BDNFおよび血管内皮増殖因子(VEGF)を含むニューロトロフィンの局所的な放出につながります。BDNFとVEGFの両方は、それぞれの受容体であるTrkBとFlk-1を刺激して、下流でAktとmTORC1を活性化するシグナルを送ります。これにより、シナプス形成とシナプス成熟を促進する様々なシナプスタンパク質の翻訳が起こります。これらの発見は、ケタミンなどのグルタミン酸作動性調節薬の迅速な効果が、従来の抗うつ薬とは異なり、活動依存的に上方制御および放出されるニューロトロフィンのプールが、別々のシナプス部位での細胞シグナル伝達の増強につながる可能性があることを示唆しています。
これらの神経栄養因子を利用するすべての戦略は、病理学的症状が損傷したニューロンおよび神経回路から生じると仮定していることを強調すべきです。この損傷は、細胞の生存だけでなく、これらのニューロンの適切な生理学的機能と接続性をも意味します。神経栄養因子によって活性化されるシグナル伝達経路の理解が深まるにつれて、新しい薬物開発を通じてこれらの経路を操作する代替戦略が考案される可能性があります。全く異なるアプローチは、組織損傷または炎症後に痛覚過敏を誘発するNGFのようなニューロトロフィンを遮断することです。NGFに対する中和抗体の開発と、痛覚閾値を下げるためのその投与は、多くの臨床的疼痛状態において有効であることが証明されています。例えば、NGFは、末梢神経系(PNS)から中枢神経系(CNS)に慢性的な変形性関節症(OA)の疼痛シグナルを送る侵害受容ニューロンを活性化することが示されています。抗NGF抗体による治療は、中等度から重度のOA患者の関節痛を抑制し、機能を改善することが示されています。神経変性疾患および精神疾患の核心的な病態生理学的メカニズムのさらなる理解は、神経栄養因子システムを関与させる合理的な治療法の開発に利益をもたらすでしょう。
FURTHER READINGS
An JJ, Liao G-Y, Kinney CE, Sahibzada N, Xu B. Discrete BDNF neurons in the paraventricular
hypothalamus control feeding and energy expenditure. Cell Metab. 2015;22:175-188.
Bai Y-Y, Ruan C-S, Yang C-R, et al. ProBDNF signaling regulates depression-like behaviors in ro-
dents under chronic stress. Neuropsychopharmacology. 2016;41:2882-2892.
Berton O, McClung CA, Dileone RJ, et al. Essential role of BDNF in the mesolimbic dopamine
pathway in social defeat stress. Science. 2006;311:864-868.
Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of
NT4/5 or BDNF. Science. 1995;267:1662-1666.
Casey BJ, Glatt CE, Lee FS. Treating the developing versus developed brain: translating preclinical
mouse and human studies. Neuron. 2015;86:1358-1368.
Castrén E, Monteggia LM. Brain-derived neurotrophic factor signaling in depression and anti-
depressant action. Biol Psychiatry. 2021;90:128-136.
Chao MV. Neurotrophins and their receptors: a convergence point for many signaling pathways.
Nat Rev Neurosci. 2003;4:299-309.
Chen ZY, Jing DQ, Bath KG, et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-
related behavior. Science. 2006;314:140-143.
Deyama S, Bang E, Wohleb ES, et al. Role of neuronal VEGF signaling in the prefrontal cortex in
the rapid antidepressant effects of ketamine. Am J Psychiatry. 2019;176:388-400.
Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen
Psychiatry. 1997;54:597-606.
Felmingham KL, Dobson-Stone C, Schofield PR, Quirk GJ, Bryant RA. The brain-derived neuro-
trophic factor Val66Met polymorphism predicts response to exposure therapy in posttrau-
matic stress disorder. Biol Psychiatry. 2013;73(11):1059-1063.
Giza JI, Kim J, Meyer HC, et al. The BDNF Val66Met polymorphism disassembles dendritic spines,
altering fear extinction circuitry and behavior. Neuron. 99(6):1356.
Je HS, Yang F, Ji Y, Nagappan G, Hempstead BL, Lu B. Role of pro-brain-derived neurotrophic
factor (proBDNF) to mature BDNF conversion in activity-dependent competition at develop-
ing neuromuscular synapses. Proc Natl Acad Sci U S A. 2012;109:15924-15029.
Lee FS, Chao MV. Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc
Natl Acad Sci U S A. 2001;98:3555-3560.
Lee FS, Kim AH, Khursigara G, Chao MV. The uniqueness of being a neurotrophin receptor. Curr
Opin Neurobiol. 2001;11:281-286.
Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneuro-
trophins. Science. 2001;294:1945-1948.
Li A, Jing D, Dellarco DV, et al. Role of BDNF in the development of an OFC-amygdala circuit regu-
lating sociability in mouse and human. Mol Psychiatry. 2021;26:955-973.
Lyons WE, Mamounas LA, Ricaurte GA, et al. Brain-derived neurotrophic factor-deficient mice
develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormal-
ities. Proc Natl Acad Sci US A. 1999;96:15239-15244.
Mariga A, Mitre M, Chao MV. Consequences of brain-derived neurotrophic factor withdrawal in
CNS neurons and implications in disease. Neurobiol Dis. 2017;97:73-79.
Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. Mechanism of TrkB-medi-
ated hippocampal long-term potentiation. Neuron. 2002;36:121-137.
Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev
Neurosci. 2013;14:7-23.
Petryshen TL, Sabeti PC, Aldinger KA, et al. Population genetic study of the brain-derived neuro-
trophic factor (BDNF) gene. Mol Psychiatry. 2010;15(8):810-815.
Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription
factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358-2361.
Shirayama Y, Chen ACH, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic
factor produces antidepressant effects in behavioral models of depression. J Neurosci.
2002;22:3251-3261.
Song M, Martinowich K, Lee FS. BDNF at the synapse: why location matters. Mol Psychiatry.
2017;22:1370-1375.
Strand AD, Baquet ZC, Aragaki AK, et al. Expression of profiling of Huntington’s disease
models suggests BDNF depletion plays a major role in striatal degeneration. J Neurosci.
2007;27:11758-11768.
Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593-598.
Van Zutphen EM, Rhebergen D, van Exel E, et al. Brain-derived neurotrophic factor as a possible
predictor of electroconvulsive therapy outcome. Transl Psychiatry. 2019;9:155.
Wise BL, Seidel MF, Lane NE. The evolution of nerve growth factor inhibition in clinical medi-
cine. Nat Rev Rheumatol. 2021;17:34-46.
Zhao M, Chen L, Yang J, et al. BDNF Val66Met polymorphism, life stress and depression: a meta-
analysis of gene-environment interaction. J Affect Disord. 2018;227:226-235.
