
- 1.11 ニューロン内シグナル伝達
- 図1.11-1:シグナル伝達の概要
- 図1.11-2:ニューロンにおけるニューロン内シグナル伝達を媒介する3つの主要な受容体タイプの概要:リガンド開口型イオンチャネル、Gタンパク質共役受容体(GPCR)、および受容体型チロシンキナーゼ(RTK)。
- 図1.11-3:Gタンパク質の機能の概要
- 図1.11-3:Gタンパク質機能の概要
- 図1.11-4:サイクリックアデノシン一リン酸(cAMP)シグナル伝達経路の基本的な構成
- 図1.11-5:ホスファチジルイノシトールシグナル伝達経路の基本的な構成
- 図1.11-6:サイクリックグアノシン一リン酸(cGMP)シグナル伝達経路
- 図1.11-7:アラキドン酸シグナル伝達経路の構成
- 図1.11-8:Gタンパク質共役受容体(GPCR)の脱感作と内在化による調節
- 図1.11-9:受容体型チロシンキナーゼを介した神経栄養因子シグナル伝達の一般的な構成
- 図1.11-10:ホスホイノシチド3-キナーゼ(PI3K)経路
- 図1.11-11:ラパマイシン標的タンパク質(mTOR)によるタンパク質翻訳の調節
- 図1.11-12:古典的なWntシグナル伝達経路の基本的な構成
- 図1.11-13:長期増強(LTP)とシナプス可塑性に関わるシナプス後メカニズムの概略図の例
- 図1.11-14:機能的選択性
1.11 ニューロン内シグナル伝達
JOHN A. GRAY, M.D., PH.D.
ニューロンシグナル伝達の概要
シグナル伝達とは、細胞が細胞外シグナルを細胞内シグナルに変換し、それに続く細胞機能の変化につながる一連の事象の連鎖のことです。シグナル伝達の最初のステップは、通常、神経伝達物質などの細胞外シグナルが、特定の細胞膜受容体に結合することを含みます。この分子(またはリガンド)がその受容体に結合すると、受容体のユニークなコンフォメーションが安定化され、その結果、細胞膜を横断してシグナルが伝達されます。最も単純な形では、神経伝達物質がイオンチャネルに結合することで、チャネルタンパク質のコンフォメーションが安定化され、チャネルが開いてイオンが細胞内外に流れ込み、一連の細胞内事象が開始されます。より複雑なシグナル伝達の例では、受容体の安定化されたコンフォメーションにより、他のタンパク質が受容体の細胞内部分に結合することができます。これらの細胞内タンパク質はその後「活性化」され、様々な下流の事象を開始します。
各シグナル伝達経路の詳細が議論される前に、受容体結合からニューロン機能の最終的な変化に至る情報伝達の共通のテーマを理解することは有用です。一般に、これらのシステムはいくつかの層に組織化されています(図1.11-1)。まず、細胞外シグナルは受容体によって検出され、細胞膜を横断してアダプタータンパク質に伝達されます。これらのアダプタータンパク質は、次に1つ以上の細胞内シグナル伝達経路を活性化し、それが、直接的またはプロテインキナーゼなどの中間体を介して、エフェクタータンパク質の機能を変化させます。プロテインキナーゼは、タンパク質にリン酸基を追加する酵素です。タンパク質のリン酸化は、リン酸化がタンパク質のコンフォメーションを変化させ、その酵素活性や他のタンパク質との結合能力を変化させることができるため、シグナル伝達における主要なメカニズムです。典型的には、タンパク質のリン酸化はタンパク質の活性化につながります。これらのシグナル伝達経路の最終的な結果は、ニューロン活動の変化と様々な遺伝子の発現の変化です。
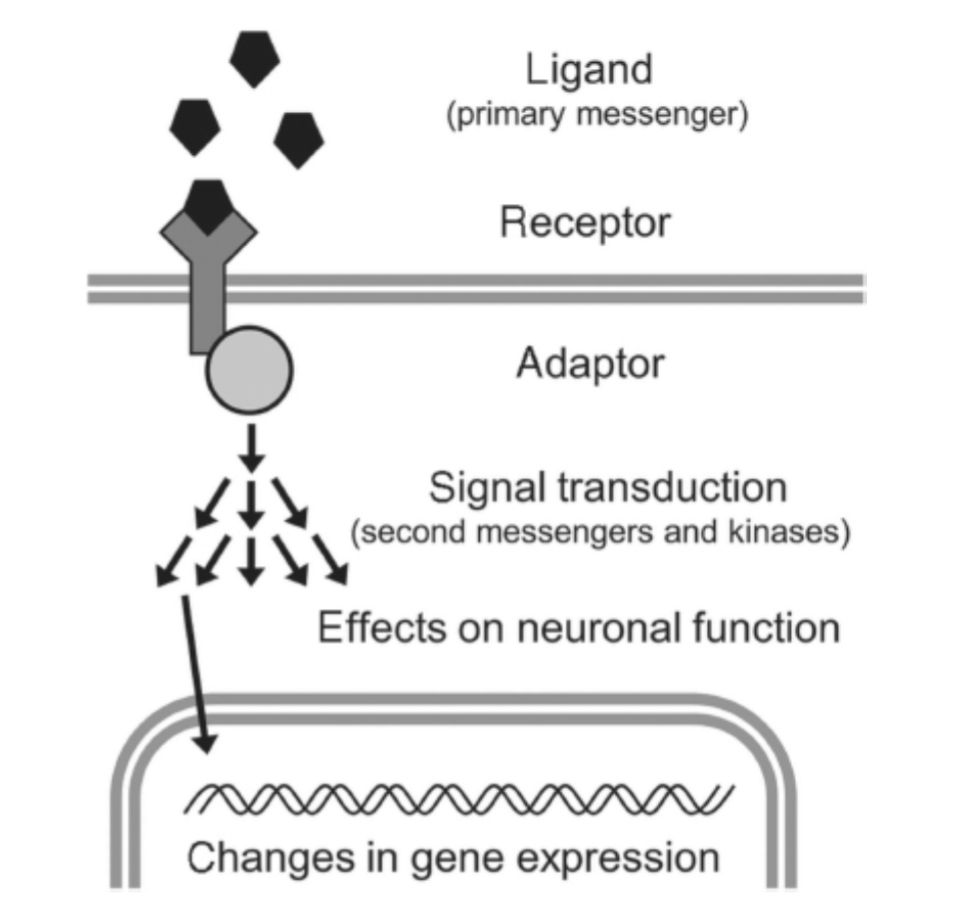
図1.11-1:シグナル伝達の概要
細胞外シグナルから細胞内シグナルへの伝達は、通常、神経伝達物質などのリガンドが受容体に結合することを含みます。受容体タンパク質のコンフォメーション変化は、シグナルを細胞膜を横断してアダプタータンパク質に伝達し、その後、アダプタータンパク質が1つ以上の細胞内シグナル伝達経路を活性化します。これらのシグナル伝達経路は、多くの場合、低分子セカンドメッセンジャーの生成やキナーゼカスケードの活性化を伴い、ニューロン機能や遺伝子発現の変化につながります。
では、なぜニューロンや他の細胞は、このような複雑な細胞内シグナル伝達経路を持っているのでしょうか?第一に、細胞外シグナルを細胞膜を横断して伝達するだけでなく、これらのシグナル伝達経路はシグナルを増幅し、細胞が非常に微量の細胞外刺激に対しても大きな応答を可能にします。さらに、細胞内シグナル伝達経路の多様性は、シグナルを特定の様式で指向させることを可能にし、それによって細胞が、適切な場合にのみ統合できる別々の情報チャネルを維持することを可能にします。例えば、これらの別々の情報チャネルは、ニューロンが異なる刺激が同時に提示されたときにそれを検出し、それに応じて応答を変化させることを可能にします。加えて、各シグナル伝達経路は、異なる種類の情報を最適に処理することを可能にする独特の空間的および時間的特性を持っています。例えば、場合によっては、ニューロンがまれな刺激に対して非常に高い感度を持つことが有利である一方で、反復的な入力は無視することが有利であるかもしれません。したがって、これらの複雑なシグナル伝達経路は、ニューロンの感度と、現在の状況と過去の経験という文脈における環境への応答性を決定します。
全体として、ニューロン内で機能する複雑な生化学的プロセスを詳細に理解することは、脳が個々の刺激にどのように応答するだけでなく、いかに無限の環境変化に適応し続けることができるかを理解するために不可欠です。さらに、ニューロン内で起こる分子プロセスに関する理解の進歩は、行動の基盤と向精神薬の作用に関する洞察を深め、改善された精神医学的治療法および診断ツールの開発を導く可能性が高いでしょう。
主要なニューロンシグナル伝達経路
ニューロンにおけるシグナル伝達には、主に3つのスキームがあります(図1.11-2)。最初のスキームはリガンド開口型イオンチャネルを伴い、これはグルタミン酸やγ-アミノ酪酸(GABA)などのアミノ酸神経伝達物質に対するシグナル伝達の主要なメカニズムです。加えて、アセチルコリンやセロトニンを含む他の多くの神経伝達物質は、リガンド開口型イオンチャネルである受容体のサブセットを持っています。神経伝達物質が結合すると、これらのイオンチャネルは特定のイオン、通常はNa⁺、K⁺、Ca²⁺、またはCl⁻に対するチャネルのコンダクタンスを変化させるコンフォメーションで安定化されます。シナプスでは、これらの受容体は細胞外シグナルを非常に迅速にシナプス後電気信号に変換し、いわゆる「速いシナプス伝達」を構成します。リガンド開口型イオンチャネルの典型的な例はニコチン性アセチルコリン受容体であり、これはアセチルコリンに対する2つの結合部位を持つ5つのタンパク質サブユニットから構成され、中央に水性の孔を囲んでいます。アセチルコリン結合部位の両方が満たされると、チャネルの内部の孔が開き、Na⁺イオンがその電気化学的勾配に従って細胞内に流入することを可能にします。Ca²⁺が細胞に入ることを可能にするチャネルは、細胞の電気的特性に影響を与え、後述するように、追加のカルシウム媒介性細胞内シグナル伝達カスケードを刺激することができます。
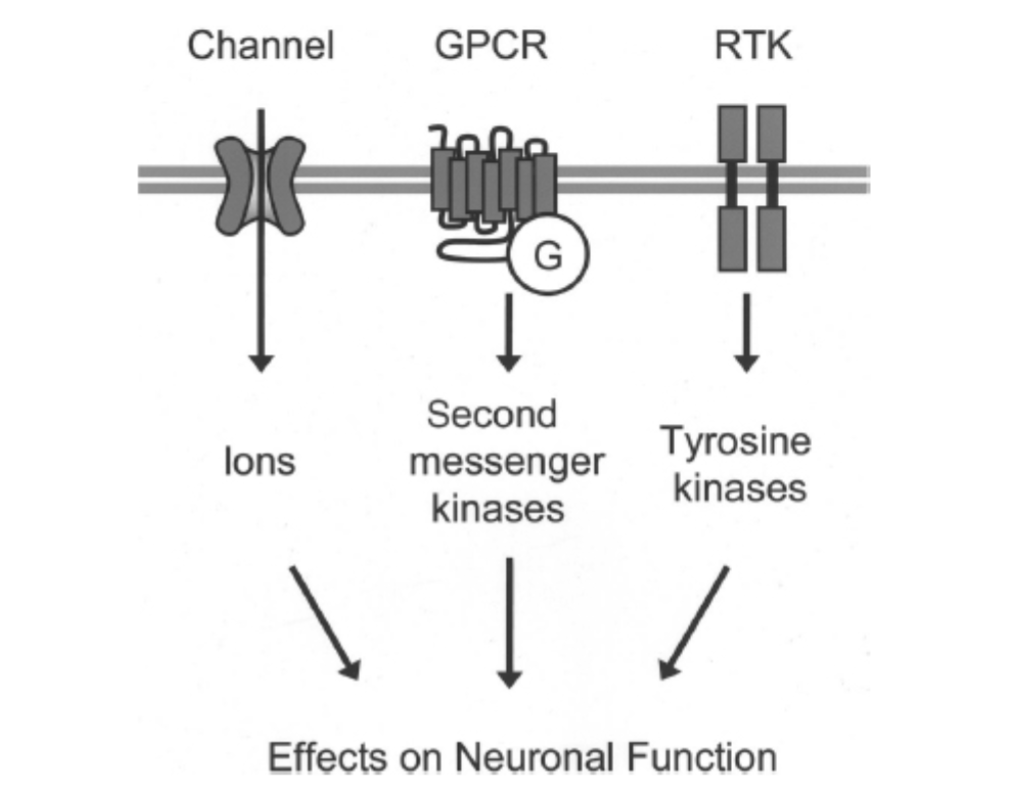
図1.11-2:ニューロンにおけるニューロン内シグナル伝達を媒介する3つの主要な受容体タイプの概要:リガンド開口型イオンチャネル、Gタンパク質共役受容体(GPCR)、および受容体型チロシンキナーゼ(RTK)。
各受容体タイプは、細胞外シグナルによって活性化されると、ニューロン機能の変化をもたらす細胞内シグナル伝達経路を誘導します。Gはヘテロ三量体Gタンパク質。
ニューロンシグナル伝達の第二の主要なスキームは、神経伝達物質が7回膜貫通型受容体に結合することを伴います。これらの受容体は、ヘテロ三量体グアニンヌクレオチド結合タンパク質(Gタンパク質)を活性化するため、GPCRsとしても知られています。GPCRsは、ゲノム内で単一の最大の受容体ファミリー(700以上のメンバーを持つ)を代表し、セロトニンやドーパミンを含む多くの神経伝達物質の主要な受容体形態です。GPCRsは主に、後に「セカンドメッセンジャー」と呼ばれる小分子を生成するエフェクター酵素を活性化するGタンパク質を活性化することによってシグナルを送ります(「ファーストメッセンジャー」は細胞外シグナルそのものです)。セカンドメッセンジャーは、次に、主にプロテインキナーゼを含む多くの下流の細胞内シグナル伝達カスケードを媒介します。小分子セカンドメッセンジャーを生成する追加のステップがあるため、GPCRsを介したシグナル伝達は、リガンド開口型イオンチャネルの開口よりも一般的に発達に時間がかかり、したがって、このスキームはニューロンにおける「遅いシナプス伝達」の大部分を占めます。
脳における第三の共通のシグナル伝達スキームは、チロシン残基のタンパク質をリン酸化する独特のクラスのプロテインキナーゼの活性化を伴います。これらのプロテインチロシンキナーゼの活性化は、NGFやBDNFなどのほとんどの神経栄養因子、ならびに様々なサイトカインやケモカインに対する主要なシグナル伝達経路です。神経栄養因子やケモカインがそれぞれの細胞膜受容体に結合すると、受容体の二量体化が起こり、別の受容体コピーが細胞膜を横断してシグナルを伝達します。これにより、プロテインキナーゼのカスケードが活性化されます。場合によっては、受容体自体の細胞内部分がカスケードの最初のチロシンキナーゼを含んでおり、他の場合には、二量体化した受容体が細胞質チロシンキナーゼをリクルートし、それが活性化されます。キナーゼのカスケードは複雑になる可能性がありますが、最終的な目的は、初期シグナルを増幅し、ニューロン機能に多数の変化を与えることです。
Gタンパク質共役受容体シグナル伝達
ゲノム中の遺伝子の少なくとも2%を占めるGPCRsは、ホルモンや神経伝達物質から匂い物質、さらには光に至るまで、幅広い分子の標的となる非常に大きなタンパク質ファミリーを構成しています。アゴニストがその同種GPCRに結合すると、受容体の活性なコンフォメーションが安定化され、受容体特異的なヘテロ三量体Gタンパク質の解離と、そのαサブユニットおよびβγサブユニットへの活性化が誘発されます(図1.11-3)。Gタンパク質はアルフレッド・ギルマン、マーティン・ロドベル、およびその同僚によって発見され、グアノシン二リン酸(GDP)とグアノシン三リン酸(GTP)の交換を分子の「スイッチ」として細胞プロセスを調節することから名付けられたタンパク質ファミリーです。ベースライン状態では、ヘテロ三量体Gタンパク質はα、β、γサブユニットから構成され、αサブユニットにGDP分子が結合しています。GPCRがアゴニストによって活性化されると、Gタンパク質αサブユニットが受容体に結合し、GDP分子を放出するコンフォメーション変化を引き起こします。
これによりGTP分子が結合し、αサブユニットが活性化されます。GTPがαサブユニットに結合すると、αサブユニットがβγサブユニットおよび受容体から解離します。これらの解離したサブユニットは生物学的に活性化され、ヌクレオチドシクラーゼ、ホスホリパーゼ、キナーゼ、イオンチャネルを含む多くの下流のエフェクターを活性化または阻害し、様々な下流の細胞効果をもたらします。このシステムは、αサブユニットが内在性GTPase活性を持ち、GTPをGDPに戻す加水分解を行うと、その基底状態に戻ります。αサブユニット内のGTPからGDPへのこの加水分解は、αサブユニットとβγサブユニットの再結合、ひいては不活性なヘテロ三量体の回復にもつながります。受容体がアゴニストに結合したままであれば、GDPは再びαサブユニットから解離し、別のGタンパク質サイクルが始まります。しかし、受容体が不活性になると、GTPからGDPへの加水分解は細胞内シグナル伝達を停止させます。
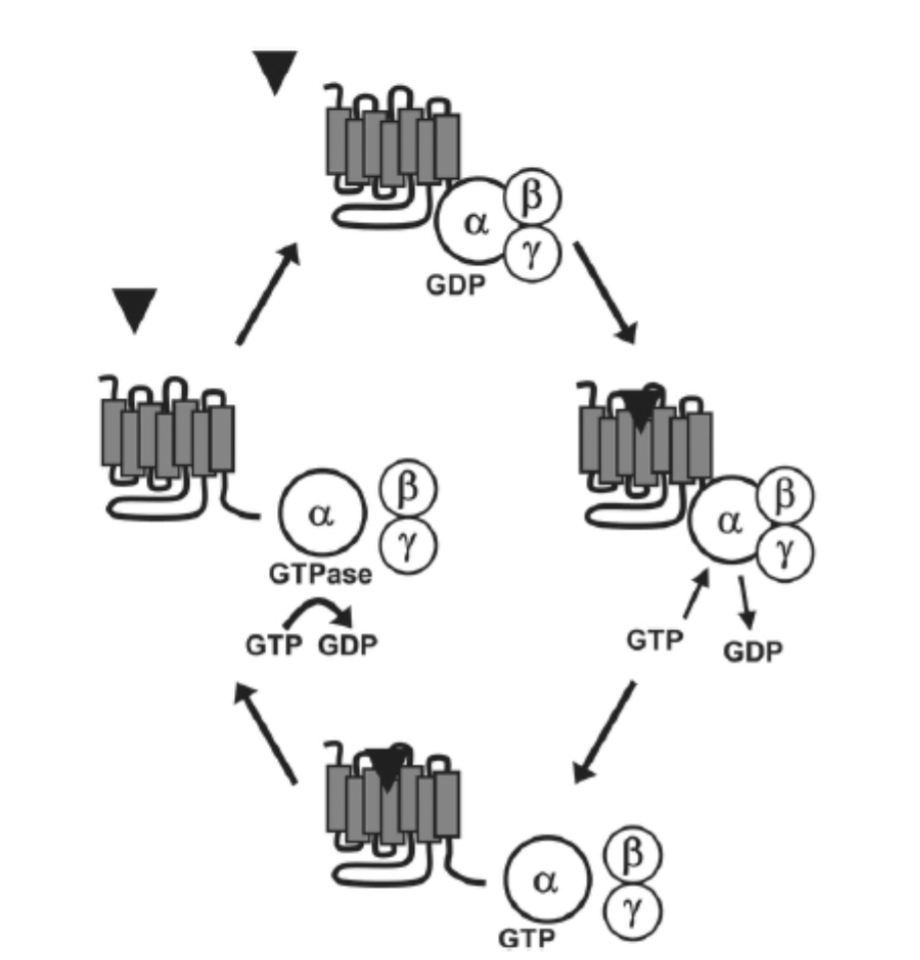
図1.11-3:Gタンパク質の機能の概要
基礎条件下では、Gタンパク質は単一のα、β、γサブユニットからなるヘテロ三量体として存在し、不活性なαサブユニットはGDPに結合しています。Gタンパク質共役受容体がそのリガンドによって活性化されると、結合しているGタンパク質がGDPを放出し、GTPの結合を可能にします。GTPがαサブユニットに結合すると、αサブユニットはβγサブユニットおよび受容体から解離します。遊離したGタンパク質サブユニットは機能的に活性であり、様々な下流のエフェクタータンパク質を活性化し、調節することができます。αサブユニットは内在性GTPase活性を持ち、GTPをGDPに加水分解し、これによりαサブユニットとβγサブユニットが再結合し、システムをその基底状態に戻します。
Gタンパク質が媒介する主要なエフェクターシステムは、共通の全体的な形態を共有しています。すなわち、活性化されたGタンパク質がエフェクター酵素を活性化し、それが小分子のセカンドメッセンジャーを生成し、その後、特定のプロテインキナーゼカスケードを開始するという形です。個々の受容体に関連する特定のGタンパク質のクラスが、どのセカンドメッセンジャーシステムが活性化されるかを決定します。神経伝達物質が関与するシグナル伝達に関わる主要な4つのGタンパク質クラスは、刺激性Gタンパク質(G_s)、抑制性Gタンパク質(G_i)、G_qタンパク質(G_q)、およびG_12/13タンパク質(G_12/13)と呼ばれ、それぞれのセカンドメッセンジャーシステムの詳細とともに以下で説明します。簡単に言うと、G_sおよびG_iのGタンパク質クラスはサイクリックアデノシン一リン酸(cAMP)システムを調節し、G_qクラスはホスホイノシチド(PI)シグナル伝達システムを調節します。G_12/13クラスはRho/Racシグナル伝達カスケードを調節しますが、ここでは議論しません。重要な点として、単一の受容体は複数のGタンパク質を活性化することができ、それらが複数のエフェクター酵素を活性化して多数のセカンドメッセンジャー分子を合成し、初期シグナルを指数関数的に増幅させることが可能です。
サイクリックアデノシン一リン酸経路
1950年代にアール・サザーランドとセオドア・ラールによってcAMPが発見されたことで、小さな細胞内分子が細胞表面受容体から細胞内の標的に情報を伝えるセカンドメッセンジャーとして機能するという概念が確立されました。この画期的な発見以来、cAMPシステムに関する数十年にわたる集中的な研究により、その動作原理が解明され、プロトタイプ的なセカンドメッセンジャーシステムとなっています。
cAMPシグナル伝達経路を活性化するGPCRは、G_sと呼ばれるGタンパク質のクラスと結合しています。G_sが受容体によって活性化されると、受容体から解離し、膜結合性のエフェクター酵素であるアデニル酸シクラーゼを刺激します。アデニル酸シクラーゼはATPをcAMPに変換します(図1.11-4)。逆に、他の受容体はG_iと呼ばれるGタンパク質のクラスと結合しており、アデニル酸シクラーゼを阻害し、cAMP産生を減少させます。したがって、特定の神経伝達物質によるcAMP産生の正味のレベルは、特定のニューロンやシナプスで発現するG_s結合型およびG_i結合型受容体サブタイプによって決まります。例えば、ノルエピネフリンはβ-アドレナリン受容体との相互作用を介してアデニル酸シクラーゼを刺激し、α2-アドレナリン受容体の刺激を介してアデニル酸シクラーゼを阻害します。
ほとんどの細胞におけるcAMPの主要な作用標的は、cAMP依存性プロテインキナーゼ、別名**プロテインキナーゼA(PKA)**であり、cAMPがニューロン機能に持つ多くの作用を媒介します。PKAは、2つの調節サブユニットと2つの触媒サブユニットからなる多サブユニットのセリン/スレオニンキナーゼです。cAMPがない場合、調節サブユニットは触媒サブユニットに結合しているため、キナーゼは不活性な状態に保たれます。しかし、cAMPが存在すると、調節サブユニットに結合し、コンフォメーション変化を引き起こして調節サブユニットを触媒サブユニットから解離させます。調節サブユニットの放出は触媒サブユニットを活性化し、それらはセリンおよびスレオニン残基の様々な細胞タンパク質を自由にリン酸化することができます。このキナーゼは、ニューロン機能のほぼあらゆる側面を調節することに関わる幅広い基質タンパク質を持っています。特に、PKAの重要なニューロン標的がいくつか特定されており、これには様々なイオンチャネル、シナプス小胞の機構、神経伝達物質合成酵素、および遺伝子転写の調節に関わるタンパク質が含まれます。したがって、cAMPレベルの変化は、幅広い時間スケールでニューロン機能に影響を与えることができます。例えば、迅速な効果はイオンチャネルの開閉や神経伝達物質放出機構を標的とすることで達成され、より緩やかな効果は神経伝達物質の合成や細胞エネルギー代謝を標的とすることで起こります。さらに、cAMPは特定の標的遺伝子の発現を制御することで、ニューロン機能に長期的な変化を引き起こします。
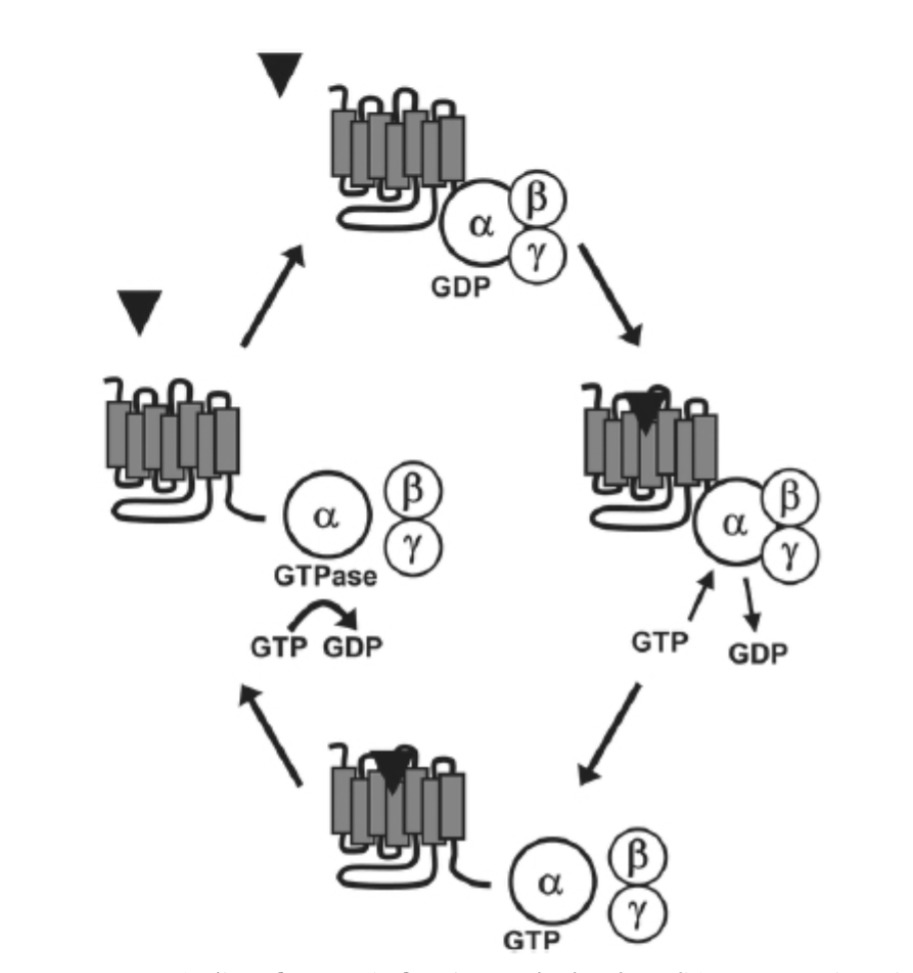
図1.11-3:Gタンパク質機能の概要
基礎状態では、Gタンパク質は単一のα、β、γサブユニットからなるヘテロ三量体として存在し、不活性なαサブユニットはGDPに結合しています。Gタンパク質共役受容体がそのリガンドによって活性化されると、結合しているGタンパク質はGDPを放出し、GTPの結合を可能にします。GTPがαサブユニットに結合すると、αサブユニットはβγサブユニットおよび受容体から解離します。遊離したGタンパク質サブユニットは機能的に活性であり、様々な下流のエフェクタータンパク質を活性化および調節することができます。αサブユニットは内在性のGTPase活性を持ち、GTPをGDPに戻す加水分解を行い、これによりαサブユニットとβγサブユニットが再結合し、システムをその基礎状態に戻します。
Gタンパク質が媒介する主要なエフェクターシステムは、共通の全体的な形態を共有しています。すなわち、活性化されたGタンパク質がエフェクター酵素を活性化し、それが小分子のセカンドメッセンジャーを生成し、その後、特定のプロテインキナーゼカスケードを開始するという形です。個々の受容体に関連する特定のGタンパク質のクラスが、どの特定のセカンドメッセンジャーシステムが活性化されるかを決定します。神経伝達物質が関与するシグナル伝達に関わる主要な4つのGタンパク質クラスは、G_s、G_i、G_q、およびG_12/13と呼ばれ、それぞれのセカンドメッセンジャーシステムの詳細とともに以下で説明します。簡単に言うと、G_sとG_iのGタンパク質クラスはサイクリックアデノシン一リン酸(cAMP)システムを調節し、G_qクラスはホスホイノシチド(PI)シグナル伝達システムを調節します。G_12/13クラスはRho/Racシグナル伝達カスケードを調節しますが、ここでは議論しません。重要な点として、単一の受容体は複数のGタンパク質を活性化することができ、それらが複数のエフェクター酵素を活性化して多数のセカンドメッセンジャー分子を合成し、初期シグナルを指数関数的に増幅させることが可能です。
サイクリックアデノシン一リン酸経路
1950年代にアール・サザーランドとセオドア・ラールによってcAMPが発見されたことで、小さな細胞内分子が細胞表面受容体から細胞内の標的に情報を伝えるセカンドメッセンジャーとして機能できるという概念が確立されました。この画期的な発見以来、cAMPシステムに関する数十年にわたる集中的な研究により、その動作原理が解明され、プロトタイプ的なセカンドメッセンジャーシステムとなっています。
cAMPシグナル伝達経路を活性化するGPCRは、G_sと呼ばれるGタンパク質のクラスと結合しています。G_sが受容体によって活性化されると、受容体から解離し、膜結合性のエフェクター酵素であるアデニル酸シクラーゼを刺激します。アデニル酸シクラーゼはATPをcAMPに変換します(図1.11-4)。逆に、他の受容体はG_iと呼ばれるGタンパク質のクラスと結合しており、アデニル酸シクラーゼを阻害し、cAMP産生を減少させます。したがって、特定の神経伝達物質によるcAMP産生の正味のレベルは、特定のニューロンやシナプスで発現するG_s結合型およびG_i結合型受容体サブタイプによって決まります。例えば、ノルエピネフリンはβ-アドレナリン受容体との相互作用を介してアデニル酸シクラーゼを刺激し、α2-アドレナリン受容体の刺激を介してアデニル酸シクラーゼを阻害します。
ほとんどの細胞におけるcAMPの主要な作用標的は、cAMP依存性プロテインキナーゼ、別名**プロテインキナーゼA(PKA)**であり、cAMPがニューロン機能に持つ多くの作用を媒介します。PKAは、2つの調節サブユニットと2つの触媒サブユニットからなる多サブユニットのセリン/スレオニンキナーゼです。cAMPがない場合、調節サブユニットは触媒サブユニットに結合しているため、キナーゼは不活性な状態に保たれます。しかし、cAMPが存在すると、調節サブユニットに結合し、コンフォメーション変化を引き起こして調節サブユニットを触媒サブユニットから解離させます。調節サブユニットの放出は触媒サブユニットを活性化し、それらはセリンおよびスレオニン残基の様々な細胞タンパク質を自由にリン酸化することができます。このキナーゼは、ニューロン機能のほぼあらゆる側面を調節することに関わる幅広い基質タンパク質を持っています。特に、PKAの重要なニューロン標的がいくつか特定されており、これには様々なイオンチャネル、シナプス小胞の機構、神経伝達物質合成酵素、および遺伝子転写の調節に関わるタンパク質が含まれます。したがって、cAMPレベルの変化は、幅広い時間スケールでニューロン機能に影響を与えることができます。例えば、迅速な効果はイオンチャネルの開閉や神経伝達物質放出機構を標的とすることで達成され、より緩やかな効果は神経伝達物質の合成や細胞エネルギー代謝を標的とすることで起こります。さらに、cAMPは特定の標的遺伝子の発現を制御することで、ニューロン機能に長期的な変化を引き起こします。
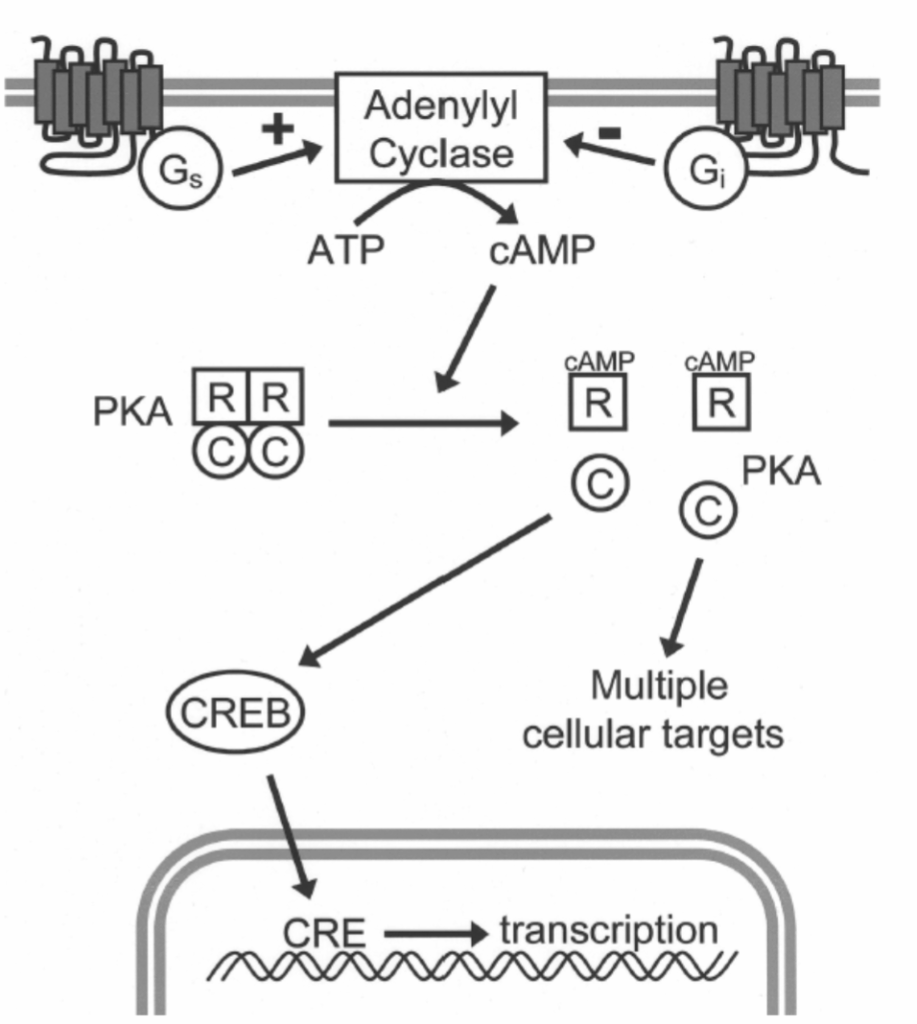
図1.11-4:サイクリックアデノシン一リン酸(cAMP)シグナル伝達経路の基本的な構成
アデニル酸シクラーゼによるATPからのcAMP産生は、Gタンパク質共役受容体によって刺激または抑制される可能性があります。G_sタンパク質に結合した受容体はcAMP合成を刺激しますが、G_iタンパク質に結合した受容体はアデニル酸シクラーゼを抑制します。cAMPが媒介する多くの細胞作用は、プロテインキナーゼA(PKA)を介して起こります。PKAは、ベースライン時には、2つの調節サブユニット(R)が2つの触媒サブユニット(C)を常に抑制する四量体として存在します。RサブユニットがcAMPに結合すると、それらはCサブユニットから解離し、活性化されて複数の細胞タンパク質をリン酸化することができます。そのような標的の1つが転写因子cAMP応答エレメント結合(CREB)タンパク質であり、これはPKAによって活性化されると、cAMP応答エレメント(CRE)と呼ばれるDNA配列に結合し、遺伝子転写を促進します。
PKAの重要な基質の一つは、cAMPレベルの上昇が遺伝子発現を調節することを可能にする転写因子です。この転写因子、CREBタンパク質は、cAMP応答エレメント(CRE)と呼ばれる短いデオキシリボ核酸(DNA)配列に結合することで、様々な遺伝子の発現を調節します。PKAによるCREBのリン酸化はそれを活性化し、標的遺伝子の制御領域にあるCRE配列に結合することを可能にし、そこで特定の遺伝子の転写を増加または減少させます。cAMPによってCREBを介して発現が調節されるタンパク質は、神経発達や生存、長期記憶の形成など、様々な神経プロセスに関与していると考えられています。
cAMPシグナル伝達の終結は、cAMPをAMPに切断する酵素である**ホスホジエステラーゼ(PDE)**の作用によって媒介されます。脳全体で発現するPDEには複数のアイソフォームがあり、これらは異なって調節されており、cAMPシグナル伝達の正確な細胞制御に複雑さを加えています。高濃度では、カフェインはPDEを非選択的に阻害し、その薬理学的効果の一部に寄与している可能性があります。さらに、個々のアイソフォームに選択的なPDE阻害剤を開発するためにかなりの努力が払われてきました。例えば、IV型PDEに選択的な阻害剤であるロリプラムは、抗うつ薬として有望でしたが、副作用のために使用が制限されました。また、脳で高レベルで発現するアイソフォームであるPDE10Aの阻害剤は、抗精神病作用を持つ可能性があります。
ホスファチジルイノシトール経路
cAMPシステムの発見後、cAMPを介して作用しない神経伝達物質受容体が多数存在することが明らかになり、他のセカンドメッセンジャーシステムの存在が示唆されました。1950年代から、PIが様々な細胞経路に関与しているというヒントはありましたが、このセカンドメッセンジャーシステムの明確な見解が浮上したのは1980年代初頭まででした。ホスファチジルイノシトールシグナル伝達経路は、cAMPシステムの多くの基本的な側面と並行していますが(図1.11-5)、いくつかのユニークな特徴も含まれています。ホスファチジルイノシトールシグナル伝達経路は、**G_qタンパク質(G_q)に結合した受容体によって開始され、このG_qタンパク質がエフェクター酵素であるホスホリパーゼC(PLC)を活性化します。この酵素は、細胞膜の細胞質側の葉に位置するイノシトール含有リン脂質であるホスファチジルイノシトール二リン酸(PIP2)**を、**ジアシルグリセロール(DAG)とイノシトール三リン酸(IP3)**という2つのセカンドメッセンジャーに切断します。これら2つのセカンドメッセンジャーは、その後、異なる細胞経路に影響を与えることができます。
疎水性であるDAGは細胞膜内に残り、そこで様々なアイソフォームのPKC(セリン/スレオニンプロテインキナーゼ)を活性化します。不活性状態のPKCは細胞質に存在しますが、DAGが生成されるとPKCは細胞膜に移動し、活性化されて複数の細胞基質をリン酸化します。水溶性のIP3セカンドメッセンジャーは細胞膜から放出され、小胞体に拡散し、そこでイノシトール三リン酸受容体に結合します。この受容体はリガンド開口型イオンチャネルであり、IP3が結合すると、小胞体からの大量のCa²⁺を急速に放出します。この放出されたCa²⁺もセカンドメッセンジャーとして機能し、様々な細胞機能を調節します。DAGに加えて、PKCの一部のアイソフォームは活性化するためにCa²⁺も必要とします。
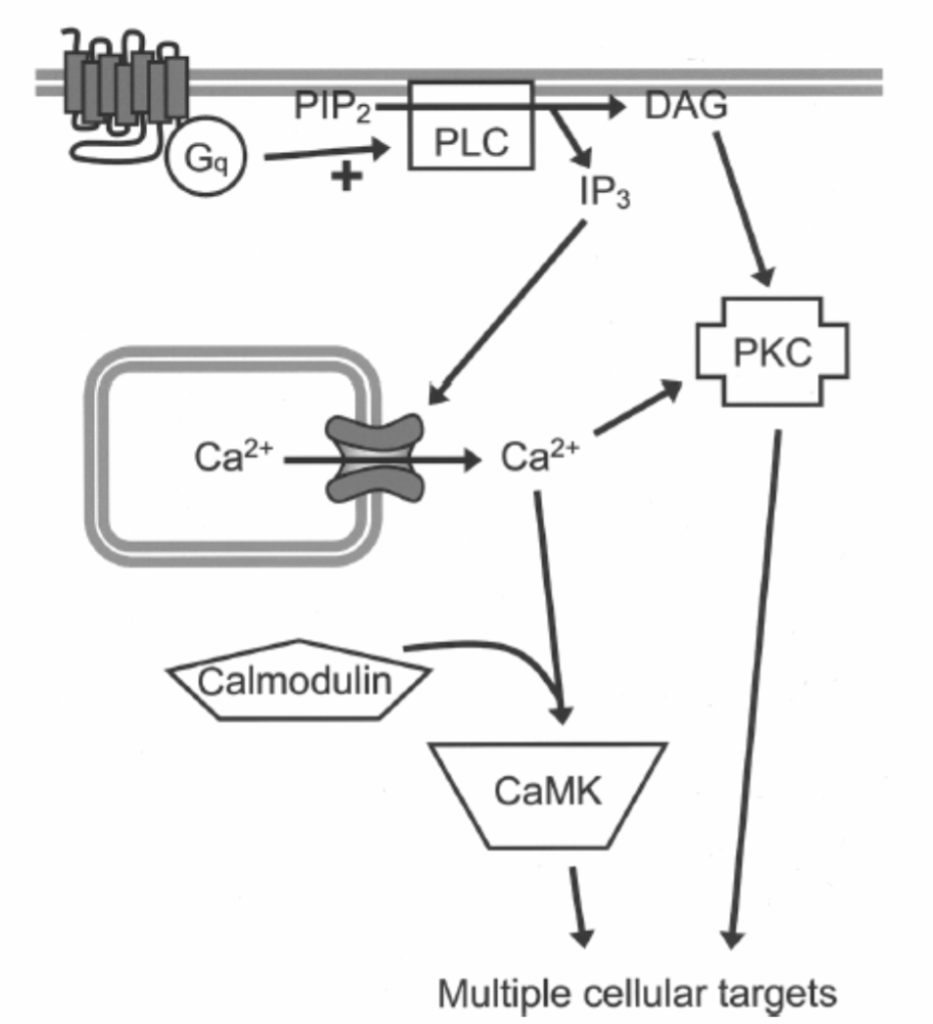
図1.11-5:ホスファチジルイノシトールシグナル伝達経路の基本的な構成
G_qクラスのGタンパク質は**ホスホリパーゼC(PLC)を活性化し、PLCは膜リン脂質であるホスファチジルイノシトール二リン酸(PIP2)を2つのセカンドメッセンジャー、ジアシルグリセロール(DAG)とイノシトール三リン酸(IP3)に切断します。IP3は小胞体に拡散し、そこでイノシトール三リン酸受容体に結合し、大量のCa²⁺を急速に放出します。このCa²⁺もセカンドメッセンジャーとして機能します。DAGはプロテインキナーゼC(PKC)**を活性化しますが、一部のアイソフォームは活性化するためにCa²⁺も必要とします。放出されたCa²⁺は、カルモジュリンに結合することでその機能の多くを媒介し、その後、カルシウム/カルモジュリン依存性キナーゼ(CaMK)を含む複数の細胞標的を活性化することができます。PKCとCaMKは、様々な転写因子を含む複数の細胞標的をリン酸化することができます。
細胞内貯蔵からのIP3誘導性のCa²⁺の急速な上昇は、ニューロン機能に即時的およびより遅延的な影響の両方をもたらします。即時的な効果は、Ca²⁺自体が様々なエフェクタータンパク質に直接結合することによって引き起こされ、シナプス小胞の放出や、細胞膜におけるカルシウム活性化イオンチャネルの開口を含みます。Ca²⁺シグナル伝達の遅延的な効果はcAMPのものと同様で、細胞エネルギー代謝や遺伝子発現への影響などがあります。カルシウムシグナル伝達のこれらのより遅い効果の多くは、カルシウムとカルモジュリン(小型で遍在性のカルシウム結合タンパク質)との結合によって媒介されます。カルモジュリンは、細胞内Ca²⁺濃度が4つのイオンがカルモジュリンタンパク質に結合するのに十分なほど高くなると活性化されます。活性化されたカルモジュリンは、カルシウム/カルモジュリン依存性(CaM)キナーゼなどの様々なキナーゼを活性化することを含む、複数の細胞標的を持っています。カルシウムに基づくシグナル伝達経路は、Gタンパク質シグナル伝達とは独立して、様々な電圧依存性およびリガンド開口型イオンチャネルによる細胞表面からのCa²⁺流入によっても活性化され得ることに注意することが重要です。
ホスファチジルイノシトール経路の終結は、複数のステップを伴います。DAGはリパーゼによってグリセロールと脂肪酸に分解されるか、膜リン脂質にリサイクルされます。Ca²⁺は、細胞膜と小胞体にあるCa²⁺-ATPaseポンプによって細胞質から迅速に除去されます。このポンプの作用は、活性化されたカルモジュリンが輸送ポンプと相互作用することによってCa²⁺自体によって強化されます。IP3はイノシトールホスファターゼによって連続的に脱リン酸化されてイノシトールになり、それが膜リン脂質に再統合されます。興味深いことに、リチウムはこれらのイノシトールホスファターゼの阻害剤であり、細胞内にIP3や他のイノシトールリン酸の蓄積につながります。これにより、さらなるシグナル伝達のために膜PIP2を補充するのに必要な遊離細胞内イノシトールの枯渇が起こり、ホスファチジルイノシトールサイクルの停止がリチウムの治療作用の根底にあるという仮説が提唱されていますが、これは依然として議論の的となっています。実際、リチウムはいくつかのアデニル酸シクラーゼやプロテインキナーゼも阻害することが知られています。
その他のセカンドメッセンジャーシステム
cAMPに加えて、もう一つの環状ヌクレオチドであるcGMPも、神経伝達物質受容体刺激によって調節されるセカンドメッセンジャーです。しかし、この2つのシステム間には顕著な違いがあります。グアニル酸シクラーゼは主に細胞質酵素であり、Gタンパク質によって直接活性化されるのではなく、**ガス状の酸化窒素(一酸化窒素)**によって活性化されます。酸化窒素は、NOSによって細胞内で合成され、NOSはカルモジュリンによって活性化されるため、細胞内Ca²⁺レベルの増加によって媒介されます(図1.11-6)。ガスがセカンドメッセンジャーとして機能するというこの実証は、細胞膜を透過して拡散でき、シナプスではシナプス前ニューロンへの逆行性シグナルとして作用する可能性があるため、細胞外メッセンジャーと細胞内メッセンジャーの区別を曖昧にします。cGMPの合成はまた、多くの場合はプロテインキナーゼGの活性化を通じて、様々な下流の効果をもたらします。cAMPと同様に、cGMPも様々なPDEによって分解されます。実際、シルデナフィル(バイアグラ)などの勃起不全治療薬は、陰茎の血管平滑筋に高度に濃縮されたPDEアイソフォームを選択的に阻害することによって作用し、細胞内シグナル伝達経路を標的とする薬剤開発の有用性を示しています。
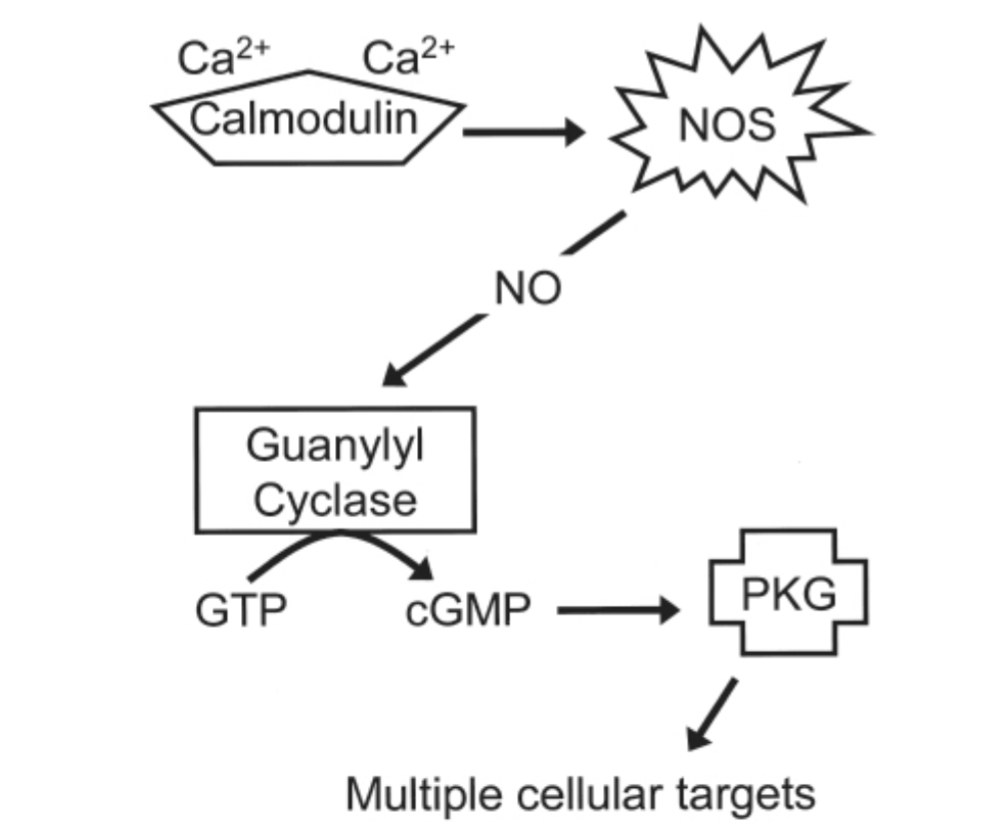
図1.11-6:サイクリックグアノシン一リン酸(cGMP)シグナル伝達経路
サイクリックアデノシン一リン酸(cAMP)システムとは対照的に、グアニル酸シクラーゼを介したcGMP合成はGタンパク質によって調節されません。代わりに、グアニル酸シクラーゼは酸化窒素によって活性化されます。酸化窒素は、カルシウム/カルモジュリン複合体によって活性化された後、**酸化窒素合成酵素(NOS)によって合成されます。cAMPと同様に、cGMPはその同種キナーゼであるプロテインキナーゼG(PKG)**を刺激することによってニューロン機能に影響を与えます。
ニューロン機能において重要な役割を果たすと思われる別の細胞内シグナル伝達システムは、脂肪酸であるアラキドン酸の代謝産物を含みます。様々な受容体がホスホリパーゼA2と呼ばれる酵素を活性化します。これはおそらく、未確認のGタンパク質または細胞質カルシウムレベルの上昇を介しています。ホスホリパーゼA2は膜リン脂質、通常はPIP2を切断し、遊離アラキドン酸を放出します。アラキドン酸は、迅速に多くの活性代謝産物に変換されます(図1.11-7)。例えば、アラキドン酸はシクロオキシゲナーゼによって切断され、複数の酵素ステップを経て、いくつかの種類のプロスタグランジンとトロンボキサンを生成する可能性があります。あるいは、アラキドン酸はリポキシゲナーゼによって切断され、ロイコトリエンを生成する可能性があります。これらの活性代謝産物は、イオンチャネルやプロテインキナーゼを含む多くの細胞内機能を調節し、アデニル酸シクラーゼやグアニル酸シクラーゼを調節することで他の経路を介したシグナル伝達を調節する上で重要です。加えて、これらの化合物は親油性であるため、ニューロンから拡散して、他のニューロン上の独自のGPCRのリガンドとして作用することができます。興味深いことに、シクロオキシゲナーゼ阻害剤は、おそらく脳内のストレス媒介性炎症プロセスを減らすことによって、統合失調症とうつ病を改善すると仮説立てられています。
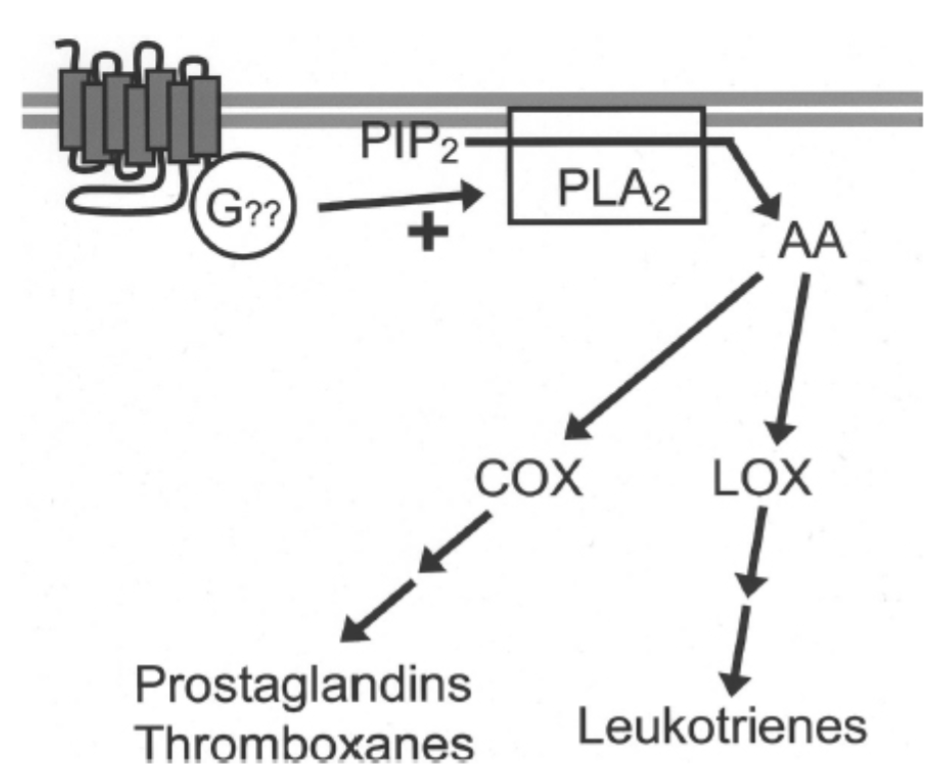
図1.11-7:アラキドン酸シグナル伝達経路の構成
様々なGタンパク質共役受容体が、おそらく未確認のGタンパク質(G??)を介して、**ホスホリパーゼA2(PLA2)と呼ばれる酵素を活性化します。PLA2は主に膜リン脂質であるホスファチジルイノシトール二リン酸(PIP2)を切断し、遊離アラキドン酸(AA)を放出します。アラキドン酸は迅速に多くの活性代謝産物に変換されます。例えば、アラキドン酸はシクロオキシゲナーゼ(COX)**によって切断されてプロスタグランジンとトロンボキサンを、**リポキシゲナーゼ(LOX)**によって切断されてロイコトリエンを生成する可能性があります。これらのAA代謝産物は、多くの細胞内機能を調節することができ、ニューロンから拡散して他のニューロン上の独自のGタンパク質共役受容体のリガンドとして作用することもできます。
GPCRによるイオンチャネルの調節
前述の通り、Gタンパク質の主要な機能は、セカンドメッセンジャーシグナル伝達カスケードを開始し、PKA、PKC、CaMキナーゼなどのキナーゼによるリン酸化を介してイオンチャネルのゲーティングを調節するなど、多数の細胞機能に影響を与えることです。さらに、cAMPやcGMPなどのセカンドメッセンジャーはイオンチャネルを直接調節することもできます。しかし、Gタンパク質自体もセカンドメッセンジャーカスケードとは独立してイオンチャネルに直接結合し、調節できることが現在明らかになっています。このプロセスは、ムスカリン性アセチルコリン受容体、α2-アドレナリン受容体、D2-ドーパミン受容体、5-HT1セロトニン受容体など、G_iに結合する受容体で最もよく確立されています。これまでと同様に、受容体活性化はGタンパク質のαサブユニットとβγサブユニットの解離を引き起こします。αサブユニットがアデニル酸シクラーゼを抑制する一方で、βγサブユニットは異なるイオンチャネルの細胞質領域に直接結合することができます。一部の細胞では、βγサブユニットはGタンパク質制御型内向き整流性K⁺(GIRK)チャネルとして知られる特定のK⁺チャネルに結合して直接開口させます。これらのチャネルは、電気化学的勾配がない場合、電流をより容易に内向きに流すため、内向き整流性と呼ばれます。しかし、通常の生理学的状況下では、GIRKを介したK⁺の流れは主に外向きであり、ニューロンの過分極につながります。他の細胞では、βγサブユニットは電位依存性Ca²⁺チャネルを直接阻害し、シナプス前小胞の放出を抑制することができます。興味深いことに、最近の証拠は、GIRKがGPCRと直接相互作用する可能性があり、Gタンパク質活性化後のイオンチャネルのほぼ瞬時の開口を促進する可能性が高いことを示唆しています。GPCRによるニューロンイオンチャネルの調節の別のメカニズムは、膜PIP2レベルの変化を介している可能性があります。多くのイオンチャネルは機能するためにPIP2を必要とするか、PIP2や他のリン脂質によって強く調節されます。したがって、これらのチャネルの活性は、G_q媒介性のPLC活性化と膜PIP2の局所的枯渇によって変化する可能性がありますが、この調節様式の生理学的関連性はまだ調査中です。
GPCRシグナル伝達の調節
GPCRが細胞シグナル伝達において極めて重要な役割を果たすため、その活動が厳密に調節されていることは驚くことではありません。実際、調節はシグナル伝達経路のほぼすべての段階で発生します。上記では、PDEによるcAMPやcGMPの分解など、セカンドメッセンジャーシグナル伝達の終結について議論しました。このセクションでは、Gタンパク質とGPCRの調節について簡単に議論します。
Gタンパク質シグナル伝達の終結は、αサブユニットの内在性GTPase活性によって媒介され、GTPをGDPに加水分解してGタンパク質を不活性化します。Gタンパク質シグナル伝達調節因子(RGS)タンパク質と呼ばれる別のクラスのタンパク質は、αサブユニットのGTPase活性を加速し、それによってGタンパク質シグナル伝達の持続時間を短縮することができます。RGSタンパク質には20以上のサブタイプがあり、脳全体で異なって発現しており、G_sファミリーを除くすべてのGタンパク質αサブユニットの調節に関与しています。RGSタンパク質の特定のサブタイプは、行動を含む重要なニューロン機能を調節することが示されています。例えば、G_qを調節するRGS2をコードする遺伝子を標的とした欠失を持つマウスは、不安の増加と雄の攻撃性の減少を示しており、行動の調節におけるRGSタンパク質の重要な役割を強調しています。興味深いことに、線条体におけるG_i媒介性のドーパミンシグナル伝達を阻害するRGS9の発現レベルが、統合失調症患者の死後脳で減少していることが判明しており、これは過剰なドーパミンシグナル伝達が精神病を引き起こすという仮説と一致しています。全体として、RGSタンパク質の多様性と不均一な分布は、個々のアイソフォームに影響を与える薬剤が高度に選択的な効果を発揮する可能性があるため、薬剤開発にとって魅力的な標的となります。
細胞内シグナル伝達カスケードを開始するだけでなく、アゴニストによるGPCRの活性化は、受容体シグナル伝達の減衰(脱感作)につながる細胞および分子メカニズムも引き起こします。脱感作は、反復的な環境刺激に対する受容体の応答性を減衰させる適応メカニズムです。全身レベルでは、受容体脱感作の根底にあるメカニズムは、オピオイドなどの精神薬理学的薬剤への耐性の発達、および抗うつ薬や抗精神病薬への治療反応の遅延の原因であると考えられています。
受容体脱感作は、通常、Gタンパク質共役受容体キナーゼ(GRK)と呼ばれるキナーゼのクラスによる受容体のフィードバックリン酸化によって媒介されます(図1.11-8)。GRKは、受容体がアゴニストに結合している場合にのみ、受容体の細胞内ドメインをリン酸化します。GRKによる受容体のリン酸化は、アレスチンと呼ばれるタンパク質が受容体に結合することを可能にし、Gタンパク質が再結合するのを防ぎ、それによって受容体を不活性にします。アレスチンのGPCRへの結合は、受容体がエンドサイトーシス小胞に内在化される原因にもなります。このプロセスは、アレスチン分子がクラスリン被覆ピットのタンパク質およびエンドサイトーシス中に切り離される膜陥入と相互作用することによって媒介されます。内在化後、受容体は細胞表面にリサイクルされるか、分解されます。受容体エンドサイトーシスの正確な役割は完全には明らかではありませんが、受容体を分解標的とすることは、慢性的な薬剤投与後の脳受容体レベルのダウンレギュレーションに関与している可能性があります。さらに、GPCRは伝統的に細胞表面からのみシグナルを送ると考えられてきましたが、最近の研究では、エンドソームを含む細胞内膜コンパートメントからの受容体シグナル伝達が示されています。細胞内コンパートメントからのGPCRシグナル伝達は、細胞膜受容体とは機能的に異なる可能性があり、薬剤はこれらの細胞内受容体に異なるアクセス性を持ちます。したがって、エンドサイトーシス膜輸送によるGPCRの調節は、新しい薬理学的薬剤の開発を可能にする可能性があります。
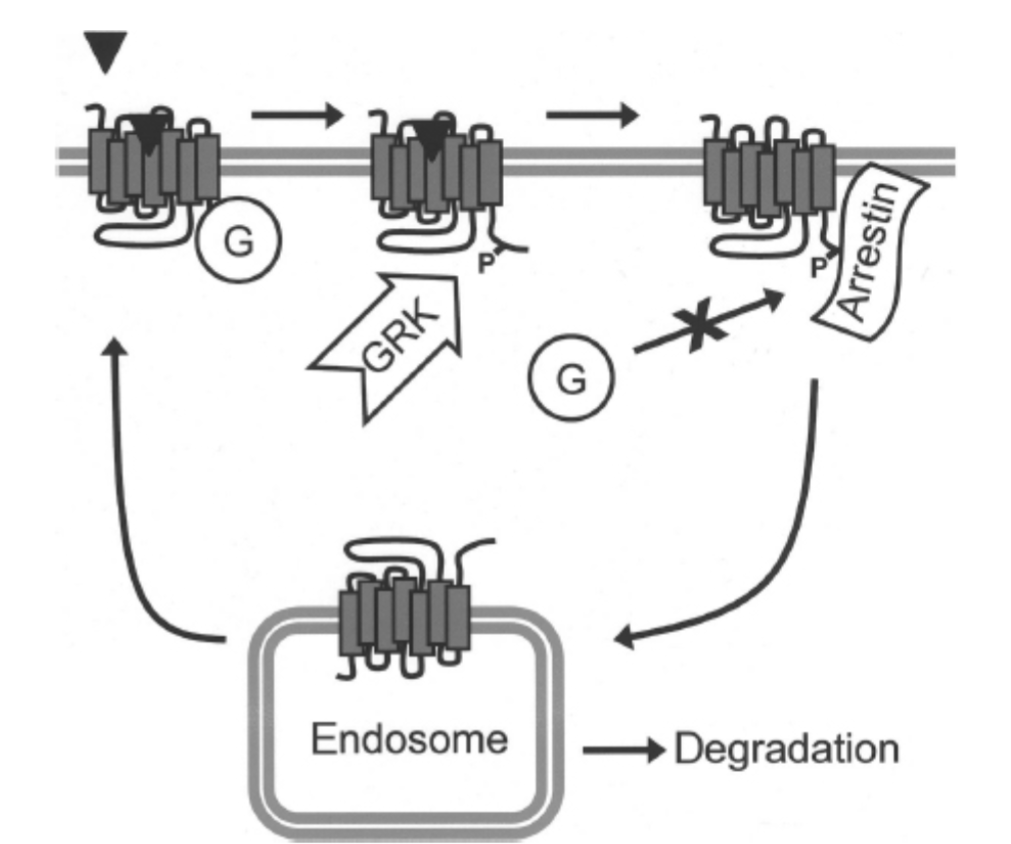
図1.11-8:Gタンパク質共役受容体(GPCR)の脱感作と内在化による調節
脱感作は、反復的な刺激に対する受容体応答性を減衰させる適応メカニズムです。GPCRの脱感作は、通常、特定の**Gタンパク質共役受容体キナーゼ(GRK)**によるフィードバックリン酸化(P)によって媒介されます。GRKによる受容体のリン酸化は、アレスチンと呼ばれるタンパク質が受容体に結合することを引き起こし、効果的にGタンパク質(G)が受容体に再結合するのを防ぎます。アレスチン結合はまた、受容体をエンドサイトーシス小胞に内在化させ、その後、受容体を細胞表面にリサイクルするか、分解を標的とすることができます。
ホスファターゼの役割
リン酸化が細胞内シグナル伝達経路において極めて中心的な役割を果たすため、プロテインキナーゼの効果を逆転させるプロテインホスファターゼも、これらのシグナル伝達経路に大きな影響を与えることは驚くことではありません。脳内に異なって分布する4つの主要なプロテインホスファターゼがあり、これらはセカンドメッセンジャーキナーゼの標的を脱リン酸化し、それぞれプロテインホスファターゼ1、2A、2B、および2Cと呼ばれます。例えば、プロテインホスファターゼ2B(カルシニューリンとも呼ばれる)は、Ca²⁺/カルモジュリンの結合によって活性化されます。したがって、G_qタンパク質に結合した神経伝達物質やCa²⁺チャネルは、カルシニューリンを活性化し、様々な細胞タンパク質のリン酸化に影響を与えることができます。実際、臓器移植拒絶反応を防ぐために使用される免疫抑制剤であるタクロリムス(Tリンパ球シグナル伝達を妨害するカルシニューリンの選択的阻害剤)によって示されるように、ホスファターゼは薬理学的薬剤の標的となり得ます。
プロテインホスファターゼを調節するもう一つの重要なメカニズムは、プロテインホスファターゼ阻害因子と呼ばれる別のクラスのタンパク質を伴います。これらのタンパク質、例えばホスファターゼ阻害因子1と2は、主要なニューロンホスファターゼであるプロテインホスファターゼ1の非常に強力な阻害因子であり、それらの阻害活性はPKAや他のセカンドメッセンジャーキナーゼによってリン酸化されると大幅に増強されます。したがって、cAMPを介してシグナルを送る神経伝達物質は、PKAの活性化と、PKA誘導性のプロテインホスファターゼ1の間接的阻害の両方によって、標的タンパク質のリン酸化に影響を与えることができます。
**ドーパミンおよびcAMP制御リンタンパク質32 kDa(DARPP-32)**と呼ばれるもう一つのプロテインホスファターゼ阻害因子は、ドーパミン入力を受ける脳領域に高度に濃縮されているため、特に興味深いものです。他のキナーゼ事象と同様に、PKAによるDARPP-32のリン酸化は、プロテインホスファターゼ1を阻害するその能力を大幅に高めます。興味深いことに、DARPP-32はカルシニューリンによって脱リン酸化され、その結果プロテインホスファターゼ1活性が増加します。カルシニューリンは細胞内Ca²⁺濃度の上昇によって活性化されるため、DARPP-32は異なる経路からの現在のシグナルの統合に関与している可能性があります。実際、DARPP-32はドーパミンシグナル伝達の主要なメディエーターであり、乱用薬物の効果において重要な役割を果たしているようです。2000年のノーベル生理学・医学賞の一部は、DARPP-32のこの役割を解明したポール・グリーングランドに授与されました。
チロシンキナーゼ経路
細胞内で起こるタンパク質のリン酸化の大部分はセリンおよびスレオニン残基に対してですが、チロシン残基のリン酸化は、異なる細胞内シグナル伝達経路において極めて重要な役割を果たします。特に、チロシンリン酸化に基づくシグナル伝達は、NGFやBDNFなどの神経栄養因子の受容体によって媒介されます。機能的には、神経栄養因子は、細胞成長と分化、シナプス可塑性、代謝、細胞生存など、幅広い細胞事象を調節するため、古典的に神経発達における役割が研究されてきました。しかし、これらの因子は全寿命にわたって発現することが示されており、新たな刺激的な研究は、神経栄養因子が行動とストレスへの応答の調節において果たす役割を明らかにしています。
プロテインチロシンキナーゼは、多様なスーパーファミリーのタンパク質を代表します。これには、細胞内ドメインにチロシンキナーゼが組み込まれた膜貫通型受容体である受容体型チロシンキナーゼと、細胞質に存在する可溶性酵素で、しばしば膜受容体にリクルートされて活性化される非受容体型チロシンキナーゼが含まれます。NGFやBDNFなどの神経栄養因子は、このセクションの主要な焦点となるTrkファミリーの受容体型チロシンキナーゼに結合します。神経栄養因子は2つの個別のTrk受容体に結合し、その結果、2つの受容体が二量体化し、それぞれの受容体の細胞質ドメインに存在するプロテインチロシンキナーゼを活性化します(図1.11-9)。活性化されたTrk受容体は、次に反対側の二量体をチロシン残基でリン酸化します。このプロセスは自己リン酸化と呼ばれます。これらのリン酸化イベントは、様々な他の細胞内シグナル伝達タンパク質のための新しい結合部位を生成します。例えば、**成長因子受容体結合タンパク質2(Grb2)**と呼ばれるアダプタータンパク質は、Trk上の特定のリン酸化チロシン残基に結合するSrc相同性2(SH2)ドメインを含み、複雑なシグナル伝達カスケードを開始します。
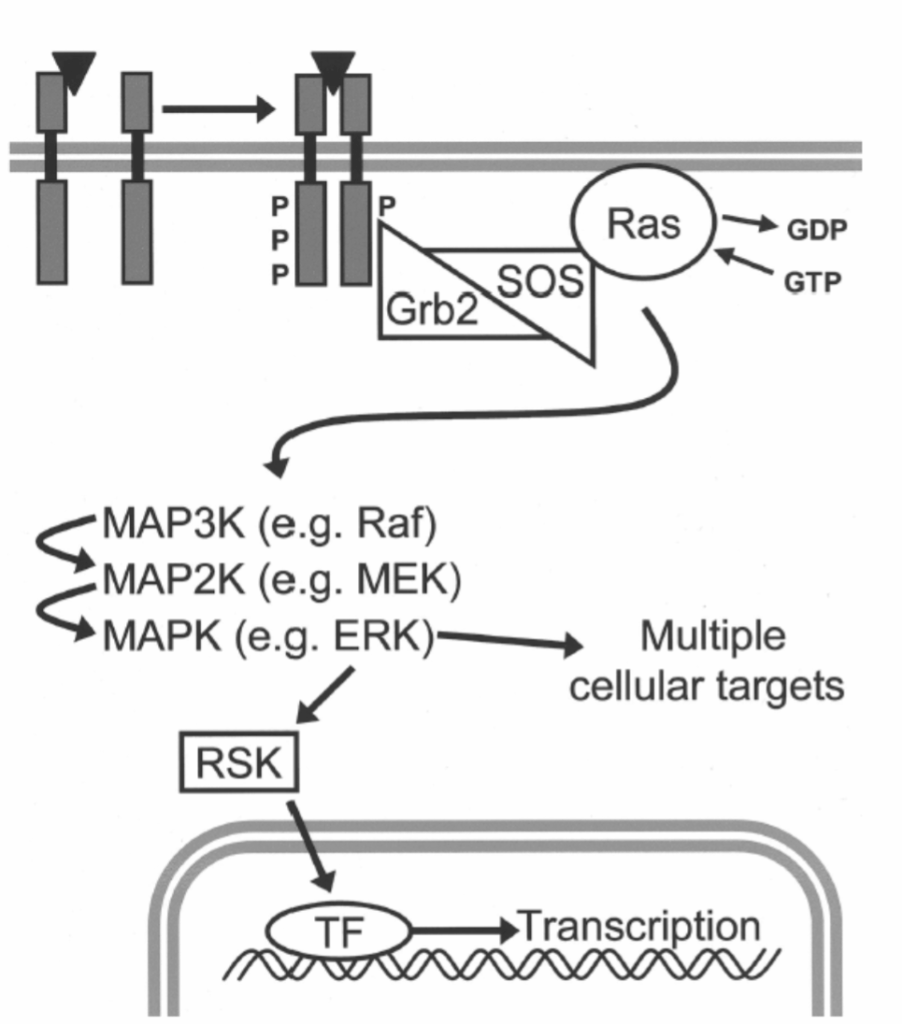
図1.11-9:受容体型チロシンキナーゼを介した神経栄養因子シグナル伝達の一般的な構成
受容体型チロシンキナーゼは、細胞内ドメインにチロシンキナーゼが組み込まれた膜貫通型受容体です。神経栄養因子の結合は、2つの受容体の二量体化と、それらの内在性チロシンキナーゼドメインの活性化と自己リン酸化を誘発します。これらのリン酸化された(P)チロシンは、成長因子受容体結合タンパク質2(Grb2)などのアダプタータンパク質の結合部位となり、その後、GDPとGTPの交換を促進することで低分子Gタンパク質Rasを活性化するSon of Sevenless(SOS)と呼ばれるタンパク質を引き寄せることができます。活性なGTP結合型において、Rasはマイトジェン活性化プロテインキナーゼ(MAPK)カスケードを含む複数の下流エフェクター経路を活性化します。細胞外シグナル調節キナーゼ(ERK)などのMAPKは、MAPK/ERKキナーゼ(MEK)などのMAPKキナーゼ(MAP2K)によって活性化され、MEKはRafなどのMAPKキナーゼキナーゼ(MAP3K)によって活性化されます。カスケードの後、ERKはp90リボソームS6キナーゼ(RSK)を含む様々な細胞標的を活性化することができ、RSKは核に移行して様々な転写因子(TF)を活性化し、遺伝子発現を調節します。
神経伝達物質受容体と同様に、Gタンパク質は活性化された受容体型チロシンキナーゼからのシグナル伝達において主要な役割を果たします。しかし、この場合、Gタンパク質はRas、Rho、およびRalファミリーのメンバーであり、総称して低分子Gタンパク質と呼ばれます。以前に説明したヘテロ三量体Gタンパク質と同様に、低分子Gタンパク質は不活性状態ではGDPに結合しており、GTPが結合すると活性化します。しかし、ヘテロ三量体Gタンパク質とは異なり、低分子Gタンパク質は受容体によって直接活性化されるのではなく、**グアニンヌクレオチド交換因子(GEF)と呼ばれる異なるタンパク質によって活性化されます。Trkの場合、Grb2がリン酸化チロシン残基に結合すると、SOS(Son of Sevenlessの略)と呼ばれるGEFタンパク質がリクルートされ、それがGDPをGTPに交換することで低分子Gタンパク質Rasを活性化します。受容体複合体にリクルートされると、SOSは多数のRas分子を活性化できるため、初期シグナルを増幅します。活性なGTP結合型において、Rasは以下に説明する複数の下流エフェクター経路を活性化することができます。RasはGTPからGDPへの加水分解によって不活性化され、これはRasGAPと呼ばれる特定のGTPase活性化タンパク質(GAP)**によって加速されます。この作用は、ヘテロ三量体Gタンパク質に対するRGSタンパク質の作用に類似しています。
マイトジェン活性化プロテインキナーゼカスケード
GPCRを介したセカンドメッセンジャーシグナル伝達経路とは対照的に、受容体型チロシンキナーゼによるRasなどの低分子Gタンパク質の活性化は、小分子中間体の産生につながりません。代わりに、低分子Gタンパク質は、3つ以上のキナーゼが連続して別のキナーゼをリン酸化するキナーゼカスケードとして組織化されたシグナル伝達経路を刺激します。いくつかの並行するキナーゼカスケードが様々な受容体によって活性化されることができ、総称してMAPK経路と呼ばれます。MAPKはセリン/スレオニンキナーゼであり、哺乳類では3つの主要なクラス(細胞外シグナル調節キナーゼ(ERK)、c-Jun N末端キナーゼ(JNK)、およびp38のアイソフォーム)に分類されます。ERK経路は、神経栄養因子や他の成長因子によって優先的に活性化される古典的なMAPK経路である一方、JNKおよびp38経路は他の受容体、シグナル伝達経路、および様々な形態の細胞ストレスによって活性化されます。
MAPK活性化につながるキナーゼカスケードは、酵母から哺乳類まで進化的に保存された組織構造に従います。MAPKキナーゼキナーゼキナーゼ(MAP4K)がMAPKキナーゼキナーゼ(MAP3K)をリン酸化し、それがMAPKキナーゼ(MAP2K)をリン酸化し、それがMAPKをリン酸化します(図1.11-9参照)。Trk受容体を介して神経栄養因子によってERKが活性化される一連の事象は、低分子Gタンパク質Rasの活性化から始まります。Rasが活性化されると、Rafと呼ばれるMAP3Kが細胞表面にリクルートされ、そこでまだ十分に記述されていないMAP4Kによってリン酸化されます。Rafは次にMEK(MAPキナーゼ/ERKキナーゼの略)と呼ばれるMAP2Kをリン酸化し、活性化します。MEKは次にERKをリン酸化し、活性化します。
ERK経路は、幅広い細胞質タンパク質や複数の転写因子を調節することに関与しているため、現在の生物医学研究においてかなりの量の研究対象となっています。例えば、ERKはp90リボソームS6キナーゼ(RSK)などのプロテインキナーゼをリン酸化し、活性化します。RSKは、c-mycやCREBを含む一連の転写因子をリン酸化します。興味深いことに、ERKシグナル伝達経路の刺激は、PKCを介して神経伝達物質受容体にも関連付けられています。したがって、ERKの活性化は長期的なニューロン機能の調節において重要な役割を果たし、他のシグナル伝達経路間の相互作用にとって重要なノードである可能性があります。
ホスホイノシチド3-キナーゼ経路
Rasの下流のシグナル伝達経路の解明は、ニューロン分化と生存に対する神経栄養因子の多くの強力な効果を媒介するもう一つの主要なキナーゼカスケードを特定しました。このカスケードには、ホスホイノシチド3-キナーゼ(PI3K)経路が関与しています(図1.11-10)。この経路では、PLCによってDAGとIP3に切断されるのと同じ膜リン脂質であるPIP2が、脂質キナーゼであるPI3Kによってリン酸化され、PIP3を生成します。PIP3はその後、様々なタンパク質を膜にリクルートする働きをします。
PIP3が膜にリクルートするタンパク質の一つに、Aktと呼ばれるセリン/スレオニンキナーゼがあります。Aktは、膜に移行すると活性化され、膜から解離し、細胞生存の制御に重要ないくつかの基質タンパク質をリン酸化します。例えば、Aktは「迅速作用型」転写因子である**核因子-κB(NF-κB)を活性化し、結果としてプロサバイバル遺伝子の転写を引き起こします。NF-κBは、阻害因子-κB(IκB)と呼ばれるタンパク質に結合した不活性状態で細胞に存在します。AktはIκBキナーゼと呼ばれるキナーゼを活性化し、IκBをリン酸化することで分解の標的とします。この分解によりNF-κBが放出され、それが核に移動して遺伝子発現を調節することができます。Aktはまた、リチウムの細胞標的である可能性がある代謝調節タンパク質であるグリコーゲンシンターゼキナーゼ3(GSK-3)**を阻害します(下記参照)。実際、現在の研究の主要な課題は、神経栄養因子の多くの効果のうち、どの効果がこれらの様々なシグナル伝達カスケードによって媒介されているかを決定することです。興味深いことに、一部のG_i結合型神経伝達物質受容体もPI3KとAktの活性化を引き起こすことができ、これらの受容体のアゴニストがニューロン生存を増強するための新規な戦略となる可能性を示唆しています。
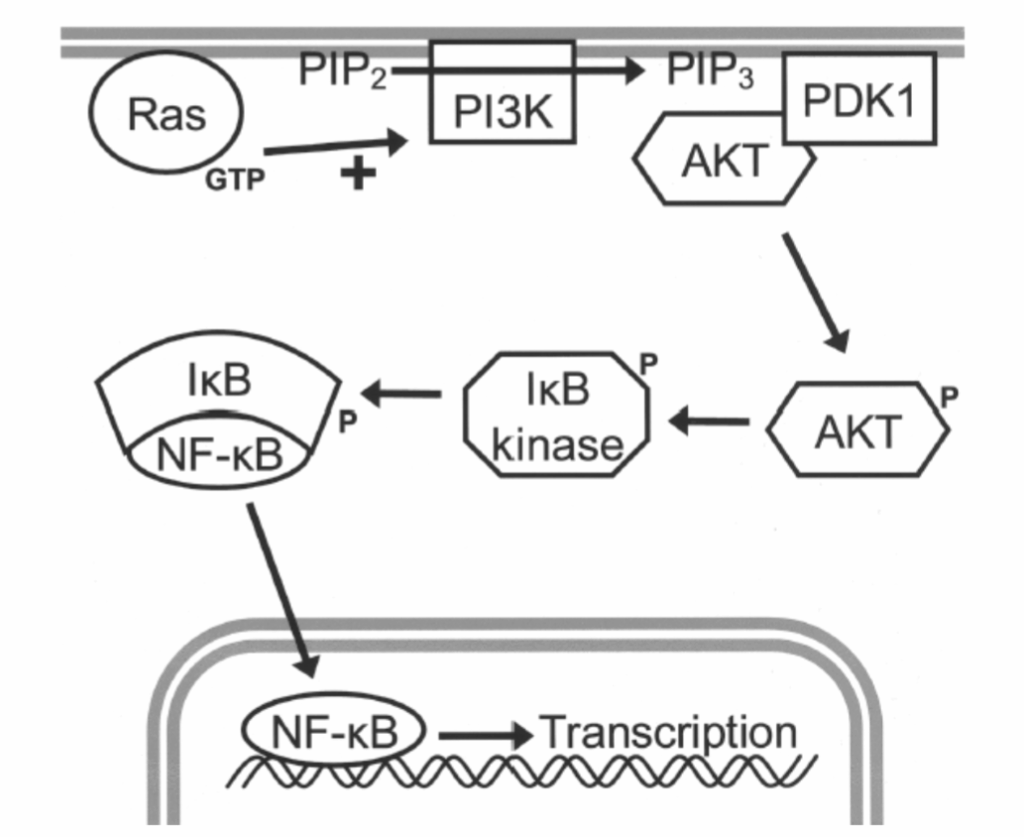
図1.11-10:ホスホイノシチド3-キナーゼ(PI3K)経路
マイトジェン活性化プロテインキナーゼ(MAPK)に加えて、受容体型チロシンキナーゼによるRasの活性化はPI3K経路を活性化することができます。PI3Kは膜リン脂質であるホスファチジルイノシトール二リン酸(PIP2)に別のリン酸を追加してホスファチジルイノシトール三リン酸(PIP3)を生成します。PIP3は、3-ホスホイノシチド依存性プロテインキナーゼ1(PDK1)やAktと呼ばれるキナーゼを含む様々なタンパク質を膜にリクルートします。PDK1はAktをリン酸化し、Aktは膜から解離して細胞生存の制御に重要ないくつかの細胞タンパク質をリン酸化することができます。例えば、Aktは核因子-κB(NF-κB)と呼ばれる転写因子の活性化につながります。NF-κBは通常、「κB阻害因子(IκB)」に結合した不活性状態で存在します。AktはIκBキナーゼと呼ばれるキナーゼを活性化し、IκBをリン酸化することで分解の標的とします。この分解によりNF-κBが放出され、その後核に移行して遺伝子発現を調節することができます。
ラパマイシン標的タンパク質(mTOR)
PI3Kの下流の標的であり、複数の細胞外シグナルの統合の中心的なノードであるのは、哺乳類ラパマイシン標的タンパク質(mTOR)と呼ばれる非典型的キナーゼです。mTORは、新しいシナプス結合の形成に必要な局所的なタンパク質合成の中心制御因子です。mTORの機能は、リガンド開口型イオンチャネル、GPCR、神経栄養因子受容体を含む主要な各クラスの受容体の活動によって影響を受けます。これは、mTORがいくつかのシグナル伝達経路の収束点におけるシグナル伝達の「ノード」として機能することを示唆しています。実際、mTOR活性は、特にPI3K-Akt経路を含む複数のシグナル伝達経路によって調節されています。
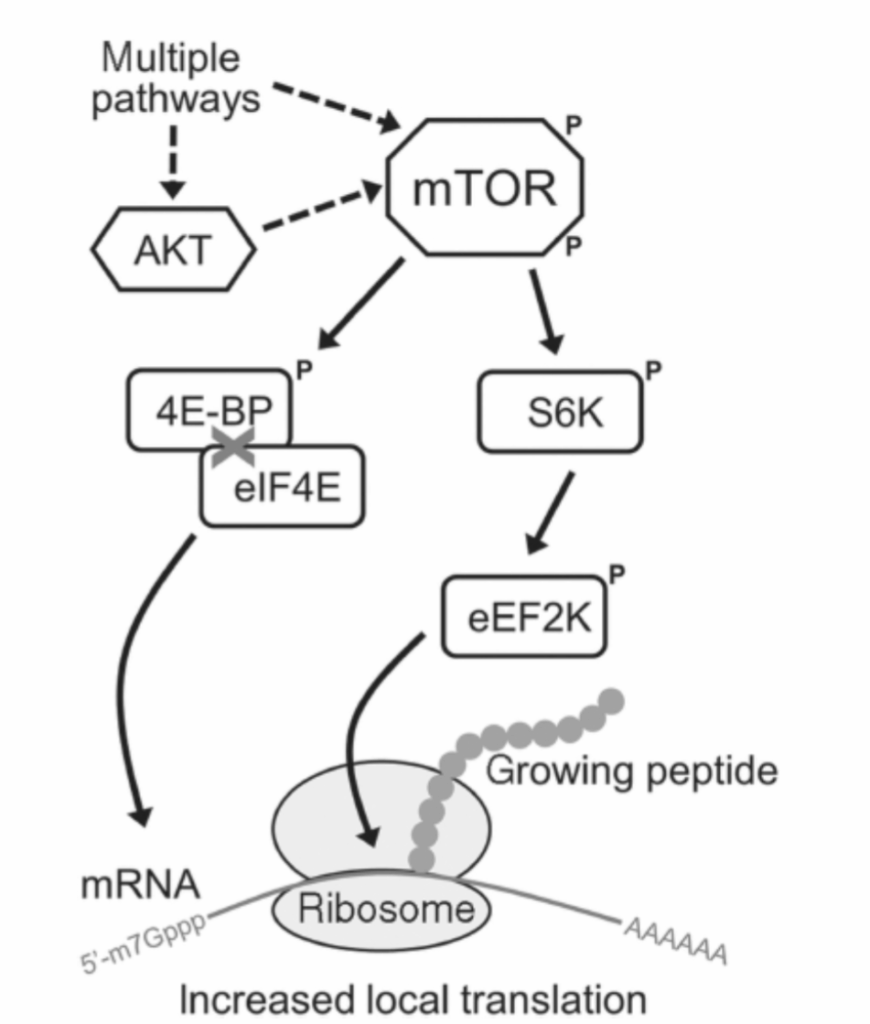
図1.11-11:ラパマイシン標的タンパク質(mTOR)によるタンパク質翻訳の調節
mTOR調節に関する研究は、mTORが複数のシグナル伝達経路、特にAktを介した重要な収束点であることを示唆しています。mTORは、リボソームにおけるタンパク質翻訳の開始および伸長に関わるタンパク質を調節することにより、局所的なタンパク質合成の中心的な制御因子です。活性化されたmTOR複合体は、4E-BP(真核生物開始因子4E結合タンパク質)をリン酸化し、eIF4E(真核生物開始因子4E)と解離させます。eIF4Eは、mRNAの7-メチルグアノシンキャップ(5′-m7Gppp)に結合することで翻訳開始を促進することができます。mTORはまた、p70リボソームS6キナーゼ(S6K)をリン酸化することで新生ポリペプチドの伸長を促進し、S6Kはその後真核生物伸長因子2キナーゼ(eEF2K)を活性化することができます。
タンパク質合成は、開始、伸長、終結の3つの一般的な段階に分けられる高度に調節されたプロセスです。mTORは、タンパク質合成の開始段階と伸長段階の両方を調節することにより、タンパク質合成のマスターコントローラーです(図1.11-11)。最初に発見されたmTOR標的の一つは4E-BP(真核生物開始因子4E結合タンパク質)でした。これは、mTORによってリン酸化されると、eIF4E(真核生物開始因子4E)を放出し、eIF4EはmRNAの7-メチルグアノシンキャップに結合することで局所的なタンパク質翻訳を開始することができます。加えて、mTORは**p70リボソームS6キナーゼ(S6K)を活性化し、S6Kはその後真核生物伸長因子2キナーゼ(eEF2K)**を活性化し、タンパク質合成を促進します。
最近、mTORシグナル伝達は、長期的なシナプス可塑性および様々な神経精神疾患と関連付けられています。例えば、ケタミンなどのNMDA受容体拮抗薬の急速な抗うつ作用は、mTORの活性化を介して、特に前頭前皮質におけるシナプスタンパク質の翻訳増加と新しいスパインの生成につながると考えられています。加えて、病的に持続するmTOR活性につながる遺伝子変異は、脆弱X症候群、自閉症、結節性硬化症を含むいくつかの神経精神疾患の根底にある可能性があります。
Wntシグナル伝達
精神医学と神経生物学において関心を集めているもう一つのシグナル伝達経路は、Wntシグナル伝達経路です。Wntは、発生において極めて重要な役割を果たすことが知られている分泌型糖タンパク質のファミリーです。しかし、Wntシグナル伝達経路の構成要素は成人脳でも発現しており、Wntシグナル伝達は成人の行動、そしておそらく精神疾患や神経疾患の病態生理において重要です。主要な、または古典的な(canonical)Wntシグナル伝達経路は、分泌型Wntタンパク質がFrizzledファミリーの細胞表面受容体に結合することから始まります。Frizzled受容体はGPCRに類似した7回膜貫通型ドメイン受容体ですが、Frizzledがヘテロ三量体Gタンパク質を介してシグナルを送るかどうかは依然として不明確です。しかし、Frizzled受容体がDishevelledと呼ばれる細胞質タンパク質を活性化し、それが最終的にβ-カテニンと呼ばれる転写共活性化因子の増加を通じて遺伝子発現の調節につながることは明らかです。
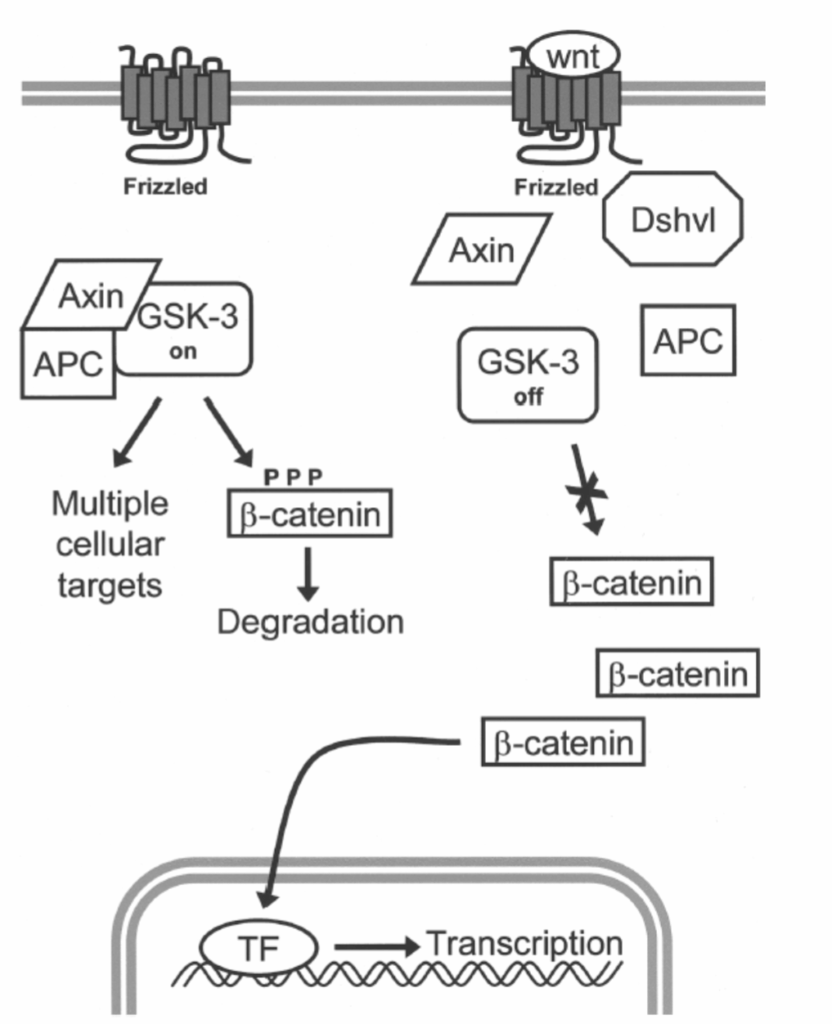
図1.11-12:古典的なWntシグナル伝達経路の基本的な構成
主要な、または古典的な(canonical)Wntシグナル伝達経路は、分泌されたWntタンパク質がFrizzledファミリーの細胞表面受容体に結合することから始まります。Wntシグナル伝達がない場合、アキシン、腺腫性ポリポーシス大腸菌(APC)タンパク質、およびグリコーゲンシンターゼキナーゼ3(GSK-3)を含むタンパク質複合体が、GSK-3を活性型(on)に維持します。GSK-3は、転写共活性化因子であるβ-カテニンを含む複数の細胞タンパク質をリン酸化します。β-カテニンがリン酸化されると、分解の標的となります。Wntシグナル伝達が開始されると、Frizzledは**Dishevelled(Dshvl)**と呼ばれるタンパク質を活性化し、それがアキシン/APC/GSK-3複合体の解離を引き起こし、結果としてGSK-3活性の抑制につながります。GSK-3活性の低下はβ-カテニンの分解を減少させ、β-カテニンは核に移行して転写因子(TF)と相互作用し、特定の遺伝子発現の変化を促進します。
Wntシグナル伝達がない場合、アキシン、GSK-3、および腺腫性ポリポーシス大腸菌(APC)タンパク質を含むタンパク質複合体が、細胞内β-カテニンレベルを調節します(図1.11-12)。GSK-3によるリン酸化を介して、このタンパク質複合体はβ-カテニンのタンパク質分解的分解を促進します。しかし、Wntシグナル伝達が開始されると、Dishevelledの活性化によりこのタンパク質複合体が解離し、他のタンパク質が結合してGSK-3活性を阻害し、β-カテニンの分解を防ぎます。これにより、細胞質β-カテニンレベルが増加し、β-カテニンは核に移行して転写因子と相互作用し、特定の遺伝子発現の変化を促進します。古典的なシグナル伝達経路に加えて、Wntシグナル伝達は細胞内カルシウムの増加やMAPK JNKの活性化を含む他の経路もたどることが示されています。したがって、Wntシグナル伝達経路の詳細は完全に解明されていませんが、他のシグナル伝達経路との間には何らかの交差および相互調節がある可能性が高く、これは活発な研究分野です。
グリコーゲンシンターゼキナーゼ3
当初はグルコース代謝の調節に関与するキナーゼとして発見されましたが、GSK-3は精神科および神経科の薬剤開発の有望な標的として浮上しています。1996年、リチウムがGSK-3を阻害することが発見され、GSK-3阻害が双極性障害の治療に役割を果たす可能性が浮上しました。近年、GSK-3の阻害が気分安定化のための治療上関連する標的であるという仮説を裏付ける研究が増加しています。
GSK-3はニューロンとグリアの両方に見られる遍在性のキナーゼであり、高度に相同ですがわずかに異なる生物学的効果を持つ2つのアイソフォームを持っています。通常、構成的に活性であると考えられており、これは、シグナルが停止を指示するまで標的タンパク質をリン酸化し続けることを意味します。例えば、上記で説明したように、GSK-3によるβ-カテニンの構成的リン酸化はタンパク質分解的分解につながりますが、Wnt経路を介したシグナル伝達はGSK-3をオフにし、β-カテニンを遊離させて遺伝子発現に影響を与えます。GSK-3は1980年にグリコーゲンシンターゼをリン酸化して不活性化する酵素として最初に特徴づけられ、インスリンシグナル伝達と糖尿病におけるその役割の研究につながりました。実際、受容体型チロシンキナーゼであるインスリン受容体へのインスリン結合は、Aktの活性化につながり、AktもGSK-3をリン酸化して不活性化します。GSK-3は通常、グリコーゲンシンターゼをリン酸化して不活性化するため、インスリンがGSK-3をオフにする能力により、細胞は上昇した血漿グルコースレベルを利用してグリコーゲンを生成することができます。
上記で説明したように、AktはBDNFなどの神経栄養因子によっても活性化されます。これらの神経栄養因子の低レベルは、うつ病やその他の神経精神疾患に関与しています。GSK-3を調節する他のキナーゼには、PKA、PKC、RSKが含まれ、GSK-3の調節メカニズムと生物学的標的が非常に多様であることを示しています。多様なシグナル伝達経路がGSK-3に収束することは、GSK-3が重要なシグナル伝達の「ノード」として位置づけられる特徴です。これらの異なる経路間のクロストークを調節する正確なメカニズムはまだ十分に確立されておらず、これは活発な研究分野です。しかし、GSK-3が細胞の異なる領域に区画化されていることが、経路間の潜在的なクロストークの多くを最小限に抑えている可能性が高いです。
シナプス可塑性
シナプスシグナル伝達の強さと効率の変化は、シナプス可塑性と呼ばれ、学習と記憶の最も重要な神経化学的基盤の一つをなします。これらのプロセスは様々な精神疾患や心理療法において重要な役割を果たすため、これらのプロセスを媒介する細胞および分子事象の定義に強い関心が寄せられてきました。シナプス可塑性は活動依存性であるため、これらの変化を調整するためには様々な細胞内シグナル伝達経路が重要です。実際、シナプス前神経伝達物質の放出の変化や、シナプス後ニューロンがそれらの神経伝達物質にどの程度効果的に応答するかの変化など、シナプス可塑性に協力的に影響するいくつかのメカニズムがあります。シナプス効率を高める主要なメディエーターとして広く考えられているシナプス後メカニズムは、**長期増強(LTP)**と呼ばれます。
LTPは、おおよそ数時間から数日間持続するシナプスの強度の増加と定義されており、脳内で記憶が形成され貯蔵される主要なメカニズムの一つとして広く考えられています。脳内のニューロン細胞型の多様性を考慮すると、LTPに関与するプロセスには多くのバリエーションがあります。しかし、LTPのプロトタイプモデルは、海馬のCA1領域のグルタミン酸作動性シナプスから導き出されています。これらのシナプスでは、α-アミノ-3-ヒドロキシ-5-メチルイソオキサゾール-4-プロピオン酸(AMPA)受容体とNMDA受容体という2つのグルタミン酸開口型イオンチャネルの作用によって開始されるLTPの初期段階と後期段階の両方が存在します。初期LTPは、シナプス内またはその近くにすでに存在するタンパク質の調節によって媒介されますが、後期LTPは新しいタンパク質合成を必要とします。
初期LTPの開始は、グルタミン酸がAMPA受容体に結合することから始まります。これによりNa⁺がシナプスに流入し、膜が脱分極します(図1.11-13)。シナプス後膜が十分に脱分極すると、NMDA受容体が開き、細胞内Ca²⁺濃度が急速に増加します。Ca²⁺レベルの上昇は、CaMキナーゼII(CaMKII)とPKCの活性化につながり、これらはAMPA受容体を急速にリン酸化し、そのNa⁺に対するコンダクタンスを高め、シナプス後応答の強度を増強します。さらに、CaMKIIの活性化は、追加のAMPA受容体のシナプス後膜への挿入を促進します。シナプスにおけるAMPA受容体の数を増やすことで、将来のシグナル刺激はより大きなシナプス後応答を生成することができます。このプロセスは、様々なGPCRを介した他のシグナル伝達経路の同時活性化によってさらに調節されることができます。
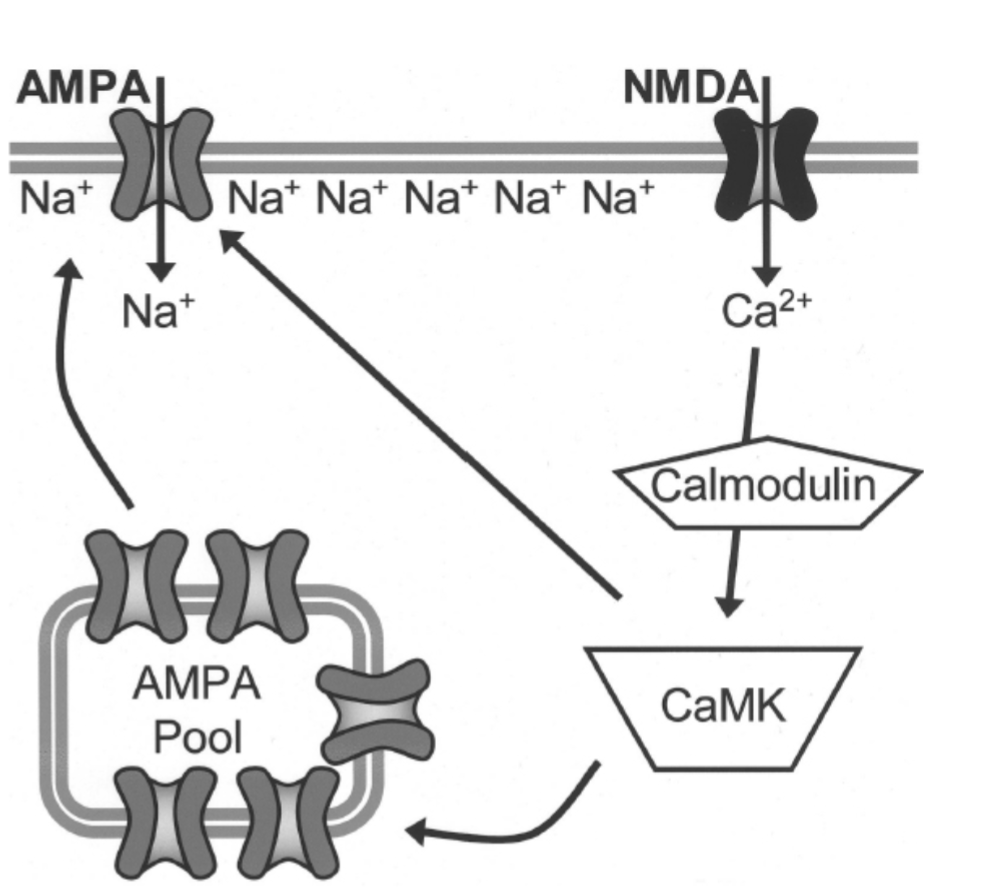
図1.11-13:長期増強(LTP)とシナプス可塑性に関わるシナプス後メカニズムの概略図の例
LTPの開始は、グルタミン酸がα-アミノ-3-ヒドロキシ-5-メチルイソオキサゾール-4-プロピオン酸(AMPA)受容体に結合することから始まります。これにより、Na⁺イオンがシナプスに流入し、膜が脱分極します。シナプス後膜が十分に脱分極すると、N-メチル-D-アスパラギン酸(NMDA)受容体が開き、細胞内Ca²⁺レベルが急速に増加します。このCa²⁺の上昇は、カルモジュリンへの結合を介して、Ca²⁺/カルモジュリン依存性キナーゼ(CaMK)とプロテインキナーゼC(PKC)を活性化します。これらの活性化されたプロテインキナーゼは、AMPA受容体をリン酸化し、さらに追加のAMPA受容体をシナプス後膜の細胞内プールに挿入するのを調節することができます。これらのプロセスが合わさって、将来のシナプス伝達の効率を高めます。
これらのリン酸化イベントは、初期LTPにおけるシナプス効率の急速な変化の根底にありますが、「後期LTP」に特徴的な永続的な変化は、新たに合成されたタンパク質がシナプスに標的化されるかどうかにかかっています。これらの新しく標的化されたタンパク質には、追加の受容体や足場タンパク質が含まれる可能性があり、シナプスのリモデリングを誘発し、刺激に対するシナプス後応答を大幅に強化することができます。この新しいタンパク質合成の少なくとも一部は、樹状突起で局所的に発生し、新しいタンパク質が活性化されたシナプスに直接標的化されることを可能にします。このプロセスは、主にmTOR活性によって調節されています。
全体として、LTPとシナプス可塑性に関わる他の細胞メカニズムは、神経科学の非常に活発な研究分野です。これらのプロセスに関する現在の理解は、シナプスシグナル伝達の厳密な時間的および空間的調節の複雑さと重要性を浮き彫りにしています。シナプス可塑性の根底にあるこれらの生化学的メカニズムの継続的な解明は、学習と行動の理解を深め、精神疾患への洞察を提供し、認知を改善できる薬理学的薬剤の開発を可能にする可能性があります。
シグナル伝達複合体と足場
シグナル伝達経路の組織化には、いくつかの追加のタイプのタンパク質が中心的な役割を果たします。これらには、足場タンパク質とアンカータンパク質が含まれます。これらは、細胞内でシグナル化されている情報が、適切かつ効率的な方法で適切な標的に伝達されることを保証するメカニズムを提供します。これらは、例えば受容体、セカンドメッセンジャー生成酵素、キナーゼ、ホスファターゼ、および基質を含む多タンパク質複合体の局所的な集合を媒介することによってこれを行います。シグナル伝達カスケードの多くの構成要素を密接に近接させることで、これらの複合体は、活性化されたタンパク質が密な細胞質を拡散して標的を見つける必要性を最小限に抑え、それによってシグナル伝達効率を大幅に向上させます。さらに、これらのシステムは、同時にシグナル伝達イベントが発生しているときに異なるシグナルの分離、または区画化を維持し、この情報の調整と統合に決定的に関与しています。これらの複合体を介してシグナル伝達経路が相互作用する多数の方法の特性評価は、活発な研究分野です。
これらの足場タンパク質とアダプタータンパク質の多くは、シグナル伝達タンパク質の輸送と局在化を媒介し、これらの特殊な多タンパク質複合体を形成するために、特定のタンパク質間相互作用を使用します。タンパク質相互作用は、他のタンパク質の特定の領域を認識して結合する役割を担うアダプタータンパク質内の異なるドメインによって形成されることがよくあります。上記で言及した例は、神経栄養因子シグナル伝達に深く関与するSrc相同性(SH)ドメインです。Grb2などのアダプタータンパク質に見られるSH2ドメインは、およそ100アミノ酸長で、リン酸化チロシン残基を含む短いアミノ酸配列に特異的に結合します。複数のタンパク質がSH2ドメインを介してこれらのホスホチロシン配列に結合することができ、その一部はその後リン酸化されて活性化され、その他は他の基質をキナーゼにリクルートするアダプターとして機能します。実際、Trkなどの活性化された受容体型チロシンキナーゼは、一連の活性化シグナル伝達分子の足場として機能します。もう一つの重要な例は、Aキナーゼアンカリングタンパク質(AKAP)と呼ばれる足場タンパク質ファミリーによる細胞内の異なる部位へのPKAの局在化です。これらのAKAPはPKAの調節サブユニットに結合し、cAMPによる活性化を待ちながら特定の基質の近くにPKAを保持します。例えば、一部のAKAPは、PKAをシナプスイオンチャネルの隣に局在させ、タンパク質の拡散による遅延を排除することで基質リン酸化の速度を大幅に向上させると考えられています。
足場はまた、シナプスにおける古典的な神経伝達物質受容体とイオンチャネルを含むシグナル伝達複合体を組織化します。これらのシステムにおけるタンパク質足場形成に関わる一般的なタンパク質ドメインは、PDZドメインです。PDZドメインはヒトの400以上のタンパク質に見られ、最後の3つのアミノ酸が一般的にS/TXV(すなわち、セリン[S]またはスレオニン[T]、その後に任意のアミノ酸[X]、その後にバリン[V]または別の疎水性アミノ酸)であるタンパク質の末端C末端セグメントにしっかりと結合しますが、他のバリエーションも存在します。PDZドメインとPDZ認識エレメントを含む多数のタンパク質を考慮すると、これらのタンパク質を含む足場は非常に大きな分子複合体を形成することができます。これらの大きな足場の最もよく知られた例は、興奮性ニューロンの**シナプス後密度(PSD)**であり、これはグルタミン酸受容体とその関連するシグナル伝達タンパク質をシナプス後膜に組織化し、シナプスのサイズと強度を決定するのに役立ちます。実際、PSDには多数のタンパク質が同定されています。イオンチャネル、GPCR、キナーゼ、ホスファターゼ、および細胞骨格タンパク質はすべて、様々なアダプタータンパク質と足場タンパク質によってPSDに標的化され、維持されています。したがって、足場タンパク質は、細胞内シグナル伝達機構の組織化、効率、および特異性における主要なプレーヤーです。
機能的選択性
ほとんどの細胞表面受容体は、古典的には単一の主要な細胞内シグナル伝達カスケードを活性化すると説明されてきましたが、上記で議論したように、ほとんどの受容体は1つまたはそれ以上の追加経路も活性化します。例えば、一部のGPCRは、ホスファチジルイノシトール経路、アラキドン酸経路、およびMAPK/ERK経路などを介してシグナルを送ることが示されています。さらに、これらの受容体の活性化は、その脱感作と内在化に関わる生化学的メカニズムを刺激します。受容体薬理学の古典的な概念では、受容体リガンドは、その受容体において完全アゴニスト、部分アゴニスト、またはアンタゴニストに分類され、この分類はその受容体のすべてのシグナル伝達経路で一貫しているとされていました。言い換えれば、もしある受容体リガンドがホスファチジルイノシトール経路を完全に活性化するならば、その受容体に関連する他のすべてのシグナル伝達および調節経路も完全に活性化すると予想されました。しかし、現在では、一部のリガンドが様々な経路間で本質的に異なるレベルのシグナル伝達を生成できるという証拠があります(図1.11-14)。この現象は、多くの場合「機能的選択性」(バイアスアゴニズムとも呼ばれる)と呼ばれます。
機能的選択性のよく認識されている形態は、リガンドが古典的なGタンパク質媒介シグナル伝達経路を活性化する能力と、受容体の脱感作およびエンドサイトーシスのためにアレスチンをリクルートする能力との間の差です。アレスチンはまた、追加のシグナル伝達経路、特にMAPK/ERKシグナル伝達の足場としても機能します。この種のバイアスアゴニズムの例は、μ-オピオイド受容体(MOR)に見られます。MORは、内因性エンケファリンペプチドやモルヒネなどのオピオイド鎮痛薬の標的です。興味深いことに、特定のアレスチンアイソフォームの標的欠失を持つマウスは、モルヒネの鎮痛効果に対する耐性をあまり発現しません。したがって、アレスチンシグナル伝達よりも鎮痛作用を媒介するGタンパク質シグナル伝達に強くバイアスをかけた新しいオピオイドを開発するための重要な努力が進行中です。耐性が低下すれば、用量増加による呼吸抑制や過量摂取のリスクが低減する可能性があります。ただし、これらのアレスチンノックアウトマウスは依然としてモルヒネ依存性を発現することに注意が必要です。したがって、バイアスのかかったオピオイドアゴニストの全体的な利益は不明のままです。実際、内因性エンケファリンに類似した真にバランスの取れたオピオイドの方が、オピオイド依存症を制限する上でより有益である可能性があります。
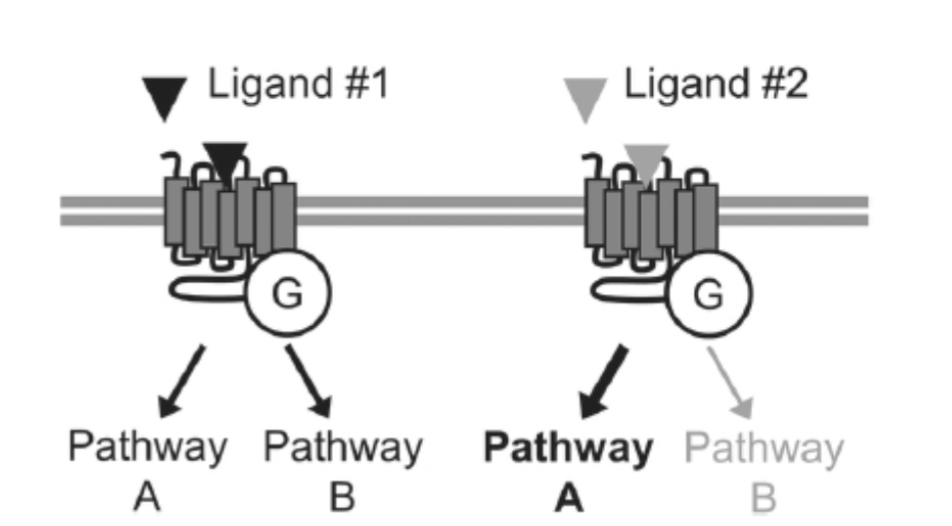
図1.11-14:機能的選択性
古典的なGタンパク質媒介シグナル伝達経路に加えて、ほとんどのGPCRは他のシグナル伝達経路も活性化できます。古典的な受容体薬理学の理論では、受容体の完全アゴニストはすべてのシグナル伝達経路を均等に活性化すると仮定していますが、現在では、一部のリガンドが本質的に様々な経路から異なるレベルのシグナル伝達を生成できることが明らかになっています。この現象は「機能的選択性」と呼ばれます。
上記の例はオピオイド受容体に焦点を当てていますが、機能的選択性は多くのGPCRで実証されており、様々なメカニズムによって媒介される可能性があります。第一に、異なるリガンドは受容体タンパク質において独自の立体構造変化をサンプリングし安定化させ、結果として様々なシグナル伝達経路の異なる活性化を引き起こします。第二に、機能的選択性は、Gタンパク質や他のシグナル伝達タンパク質および足場タンパク質の多様性、あるいはGPCRの二量体化およびオリゴマー化の観察された能力(その機能は依然として十分に理解されていない)を通じて生じる可能性もあります。第三に、上記で議論したように、薬剤は細胞内膜上の受容体と異なって相互作用する可能性があり、細胞内バイオアベイラビリティが局所的に偏った機能的選択性の形態につながる可能性を示唆しています。したがって、機能的選択性の概念は興味深いだけでなく、将来の精神科薬剤開発に重要な影響を与える可能性が高いです。
細胞内シグナル伝達のリモート制御
近年、新興の合成生物学技術により、空間的および時間的精度を伴う細胞内シグナル伝達経路の標的活性化が可能になりました。これらの技術は総称してケモジェネティクスと呼ばれ、複雑な回路と行動の研究を促進し、脳機能の理解に革命をもたらしています。一つのアプローチでは、光活性化ロドプシンの細胞内ループを特定のアドレナリン作動性受容体またはセロトニン作動性受容体のそれらに置き換えたキメラ受容体が設計されました。これらの人工受容体は「オプト-XR」と呼ばれ、光を使用して細胞内ループに使用された受容体によって通常リクルートされるシグナル伝達経路を活性化することを可能にします。光操作の速度のため、このアプローチは細胞内シグナル伝達の迅速な刺激と、緊張性活性化と相性活性化の効果の検討を可能にしますが、脳への光ファイバーの侵襲的な挿入が必要です。別の方法は、内因性受容体を活性化しない合成リガンドによってのみ活性化されるように特別に設計されたGPCR、またはケモジェネティックアクチュエーターを使用します。DREADDs(designer receptors exclusively activated by designer drugs)またはRASSLs(receptors activated solely by synthetic ligands)と呼ばれるこれらの技術は、同様に生体内での細胞内シグナル伝達の正確な時空間制御を可能にします。これらのケモジェネティックアクチュエーターの様々なバージョンは、主要なGPCRシグナル伝達経路(G_s、G_i、およびG_q)を活性化します。設計された酵素や薬剤誘導性の二量体化または不安定化ドメインを含む、細胞内シグナル伝達カスケードを制御できる追加のケモジェネティックアクチュエーターが開発されています。薬剤によって活性化されるため、ケモジェネティックアクチュエーターは細胞内シグナル伝達の非侵襲的変調を可能にし、したがって、多数の神経精神疾患に対する革新的な治療アプローチとして使用される可能性があります。
将来の方向性
分子神経生物学における進歩を診断および治療能力の向上に結びつけることは、現代精神医学が直面する最大の機会であり課題です。精神疾患の治療に使用される現在の薬剤は、細胞表面受容体が媒介する細胞間シグナル伝達の理解における数十年にわたる進歩を促進してきました。しかし、これらの受容体によって活性化される細胞内シグナル伝達経路の理解における劇的で継続的な進歩は、他の医学分野で達成されてきたように、精神疾患に対する新規で革新的な、そして改良された薬理学的薬剤につながる可能性が高いです。したがって、薬剤発見における将来の努力は、受容体レベルでのシナプス神経伝達のみを標的とする現在の戦略を超えて、細胞内シグナル伝達経路の構成要素に作用する薬剤の開発へと移行すべきです。本質的に、シグナル伝達経路はかなりの冗長性と相互作用を持っています。したがって、これらのネットワーク内の重要なポイントを特定し、標的とすることで、分子診断テストと治療の改善につながる可能性があります。
FURTHER READINGS
Airan RD, Thompson KR, Fenno LE, Bernstein H, Deisseroth K. Temporally precise in vivo control
of intracellular signalling. Nature. 2009;458:1025-1029.
Alberts B, Heald R, Johnson AD, Morgan D, Raff M, Roberts K, Walter P, Wilson J, Hunt T. Molecular
Biology of the Cell. 7th ed. W.W. Norton & Company; 2022.
*Blitzer RD, Iyengar R, Landau EM. Postsynaptic signaling networks: cellular cogwheels under-
lying long-term plasticity. Biol Psychiatry. 2005;57:113-119.
Boeckers TM. The postsynaptic density. Cell Tissue Res. 2006;326:409-422.
Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. u-Opioid receptor desensitization by
ß-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720-723.
Cheyette BN, Moon RT. Wnt protein family. In: Henry HL, Norman AW, eds. Encyclopedia of Hor-
mones. Academic Press; 2003:655-674.
Doupnik CA. GPCR-Kir channel signaling complexes: defining rules of engagement. J Recept Sig-
nal Transduct Res. 2008;28:83-91.
Duman RS, Voleti B. Signaling pathways underlying the pathophysiology and treatment of de-
pression: novel mechanisms for rapid-acting agents. Trends Neurosci. 2012;35:47-56.
Duncan AL, Song W, Sansom MSP. Lipid-dependent regulation of ion channels and G protein-
coupled receptors: insights from structures and simulations. Annu Rev Pharmacol Toxicol.
2020;60:31-50.
Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-
coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107-144.
Gillis A, Kliewer A, Kelly E, et al. Critical assessment of G protein-biased agonism at the u-opioid
receptor. Trends Pharmacol Sci. 2020;41:947-959.
Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its
potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537-568.
Herring BE, Nicoll RA. Long-term potentiation: from CaMKII to AMPA receptor trafficking. Annu
Rev Physiol. 2016;78:351-365.
Irannejad R, Pessino V, Mika D, et al. Functional selectivity of GPCR-directed drug action through
location bias. Nat Chem Biol. 2017;13:799-806.
Kroeze WK, Sheffler DJ, Roth BL. G-protein-coupled receptors at a glance. J Cell Sci.
2003;116:4867-4869.
Le Novère N, Li L, Girault JA. DARPP-32: molecular integration of phosphorylation potential. Cell
Mol Life Sci. 2008;65:2125-2127.
Müller N, Schwarz MJ. COX-2 inhibition in schizophrenia and major depression. Curr Pharm Des.
2008;14:1452-1465.
*Nestler EJ, Hyman SE, Malenka RC. Molecular Neuropharmacology: A Foundation for Clinical
Neuroscience. 4th ed. McGraw-Hill Education; 2020.
*Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neuro-
biol. 2001;11:272-280.
Peineau S, Bradley C, Taghibiglou C, et al. The role of GSK-3 in synaptic plasticity. Br J Pharmacol.
2008;153(Suppl 1):S428-S437.
*Sternson SM, Roth BL. Chemogenetic tools to interrogate brain functions. Annu Rev Neurosci.
2014;37:387-407.
*Urban JD, Clarke WP, von Zastrow M, et al. Functional selectivity and classical concepts of quan-
titative pharmacology. J Pharmacol Exp Ther. 2007;320:1-13.
Wild AR, Dell’Acqua ML. Potential for therapeutic targeting of AKAP signaling complexes in ner-
vous system disorders. Pharmacol Ther. 2018;185:99-121.
Willars GB. Mammalian RGS proteins: multifunctional regulators of cellular signalling. Semin
Cell Dev Biol. 2006;17:363-376.
Wootten D, Christopoulos A, Marti-Solano M, Babu MM, Sexton PM. Mechanisms of signalling
and biased agonism in G protein-coupled receptors. Nat Rev Mol Cell Biol. 2018;19:638-653.
Yaffe M, Cantley LC. Grabbing phosphoproteins. Nature. 1999;402:30-31.
