
1.18 ゲノム、トランスクリプトーム、プロテオーム:精神疾患の神経生物学を支える分子遺伝学、生化学、およびマルチオミクス
エヴァン・E・アフシン医学博士 および クリストファー・E・メイソン博士
細胞のDNA、リボ核酸(RNA)、およびタンパク質の構成要素に関するグローバルな研究が、実現可能でアクセスしやすく、ますます日常的になっている時代です。100万個以上の一般的な遺伝マーカーを対象としたゲノムワイド関連解析(GWAS)は、現在では比較的安価で日常的に実施されています。エクソーム、すなわち発現遺伝子のDNA、ならびに全ゲノムの配列決定に関する研究についても同様のことが言えます。国際HapMapコンソーシアムによって提供されたゲノム多様性に関する理解の深化と、遺伝子型決定技術の継続的な大きな進歩は、詳細な表現型特性と環境曝露に関する情報を持つ多数の個人において、ハイスループットで費用対効果の高いGWASを実施することを可能にしました。その結果得られたデータは、精神疾患に潜在的に関連する遺伝子変異を特定し、大規模で多様なサンプルにおけるこれらの変異の有病率を評価し、遺伝子-疾患関係の可能な修飾因子を検討するために使用されます。まもなく、1000ゲノムプロジェクトが続き、SNPs、コピー数変異(CNVs)、挿入欠失(indels)、および大規模な構造再配列を含む包括的なデータベースが構築されました。ENCODEプロジェクト(本章で後述)は、これらの研究をさらに機能ゲノミクスへと拡大しました。機能ゲノミクスは、脳研究においてすでに日常的になりつつあり、精神疾患を持つ個人の死後脳や、臨床神経科学に関連する動物モデルの研究に応用されています。
これらの画期的な分子遺伝学的アプローチは、生物学的情報をグローバルな視点から研究することを可能にします。この情報は、ヒトゲノム、すなわちヒト細胞内のDNAの全補体からなる30億塩基対の配列に収められています。各個人のゲノムは、核を持つ体内のすべての細胞に含まれており、正常な発達を可能にするために必要な原材料を表しています。この遺伝物質はまた、精神疾患を含む多くのヒト疾患において、原因的または寄与的な役割を果たしています。ゲノミクスとは、生物の遺伝物質の全補体を研究する学問です。この観点からヒトを考察する能力は比較的新しい発展であり、ヒトゲノムの配列決定とそれに続くヒトの遺伝的多様性およびすべての機能的・制御的要素を特定・特徴づけるための大規模な努力によって活性化されました。トランスクリプトームとは、生物内の遺伝子から転写されたRNAの総体を指す用語であり、このプロセスはしばしば遺伝子「発現」と呼ばれます。遺伝子発現の研究は通常、遺伝子とタンパク質の中間体であるメッセンジャーRNA(mRNA)に焦点を当てています。また、実験デザインによって異なり、成熟したポリアデニル化mRNAのみ、または細胞内の総RNA(mRNAとncRNAを含む)を含む場合があります。遺伝子発現のグローバルな研究は機能ゲノミクスの分野を構成し、ゲノムDNAまたはRNA由来の短いリードの大規模並列配列決定の力に依存する手法である**次世代シーケンシング(NGS)**などのツールを使用する場合があります。NGSのワークフローの簡略化された図を図1.18-1に示します。
プロテオームとは、生物内で発現するすべてのタンパク質を指し、プロテオミクスによってグローバルに研究されます。ヒトや他の多くの生物におけるこれらの生物学的領域の一般的な理解が深まるにつれて、その複雑性に対する認識も高まっています。その構造、機能、複雑な相互作用を深く掘り下げれば掘り下げるほど、基本的な概念を再考せざるを得なくなります。例えば、一見正常で健康な人における遺伝的多様性の量は過小評価されています。DNAの配列が個人間でわずかに異なることは長らく認識されていましたが、最近ではヒトゲノムの実に20%にも大規模な構造多様性が存在し得ることが判明しました。遺伝コードの数十万から数百万のヌクレオチドが、明らかな悪影響なしに、単一の個人内で欠失したり、重複したり、複数のコピーを持ったりする可能性があります。これらの変異はコピー数多型(CNPs)またはCNVsと呼ばれ、カタログ化され詳細に研究され始めたばかりです。さらに、単一ヌクレオチドレベルで個人間に非常に多数の一般的な違いがあり、これらはSNPsとして知られています。これらの多様な形態の遺伝的変異の重要性を理解することは、正常な発達における遺伝的構造多様性の役割に関する現在の考え方に革命をもたらし、精神疾患に関連する特性の遺伝性とその生物学的側面を解明し、最終的には精神科診断の枠組みを再構築する可能性を秘めています。
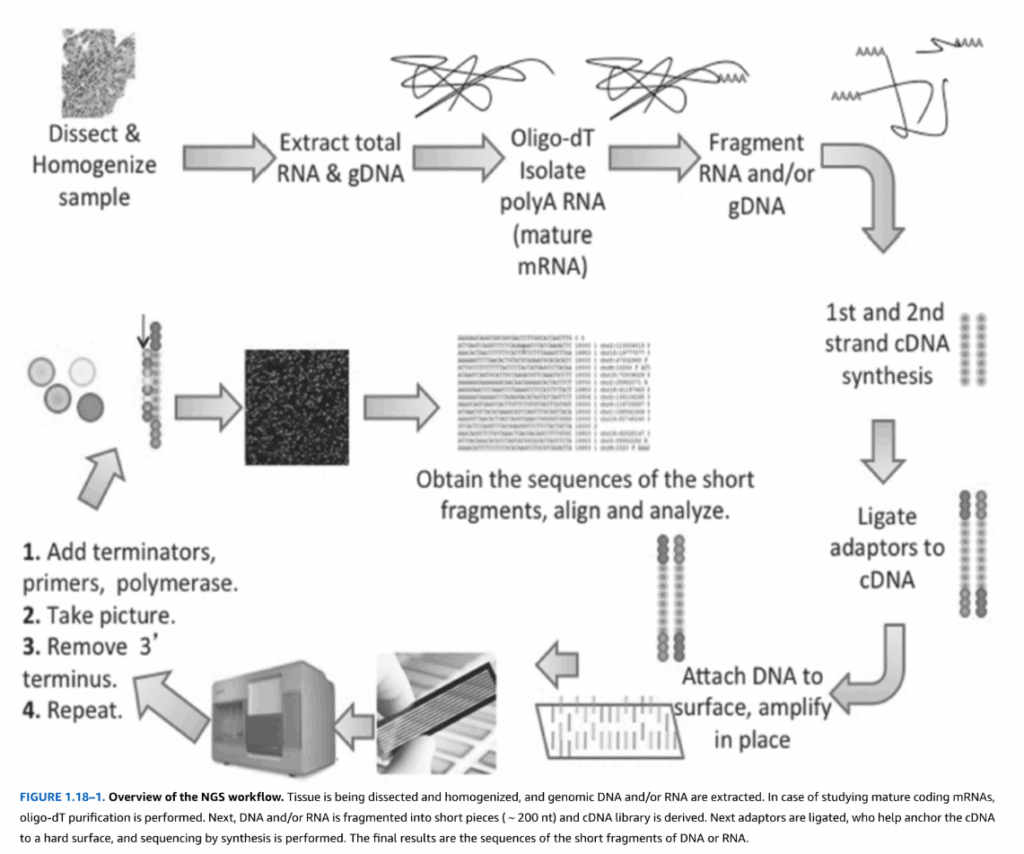
図1.18-1. NGSワークフローの概要
この図は、**次世代シーケンシング(NGS)**の一般的なワークフローを示しています。
- まず、組織が解剖されホモジェナイズされ、ゲノムDNAおよび/またはRNAが抽出されます。成熟したコード性mRNAを研究する場合は、オリゴdT精製が行われます。
- 次に、DNAおよび/またはRNAは短い断片(約200ヌクレオチド)に断片化され、cDNAライブラリが作成されます。
- 次に、cDNAを硬い表面に固定するのに役立つアダプターが結合され、合成によるシーケンシングが実行されます。
- 最終的な結果は、DNAまたはRNAの短い断片の配列です。
「遺伝子」という概念も進化しています。かつては、単一の遺伝子が単一のタンパク質の生産につながると考えられていました。しかし、1977年には、1つの遺伝子が機能的に異なる複数のバージョンのタンパク質をコードすることが発見されました。最近のデータは、個々の遺伝子の境界に関する従来の定義に異議を唱えており、これらの発見は、ゲノムの機能と調節に関する大幅な再評価につながる可能性が高いです。例えば、ほとんどの遺伝子には複数の3’末端と5’末端、様々なスプライシング部位があり、その遺伝子ボディ内に様々な制御要素を宿しています。さらに、エンハンサーと呼ばれる要素は遺伝子発現を長距離で制御でき、**転写因子結合部位(TFBSs)**は遺伝子が活性化、減衰、またはサイレンシングされる能力を調節できます。
基本的な遺伝学の概念の再考を促すだけでなく、ヒトゲノム全体の配列決定は、遺伝子多様性が遺伝子発現の調節をどのように変化させるか、また遺伝子の機能的または調節的ドメインの多様性が脳の構造と機能にどのように影響するか、そしてこの多様性が精神疾患のリスクにどのように寄与するかについての、はるかに深い理解のための舞台を整えました。
本章では、DNA、染色体、RNA、タンパク質の性質、およびコーディング遺伝子と非コーディング遺伝子の概念を含む基本的な概念をレビューします。次に、ヒトゲノムの配列特性に関する現在の知識をグローバルなスケールでレビューし、この配列が個人間および集団間でどのように異なるかを考察します。その後、ゲノムがどのように調節されるか、およびプロテオミクスとシステム生物学の研究が、機能ゲノミクスのこれらの最近の進歩をどのように活用して、正常および病的脳の発達と機能に関する一般的な理解を深めるかについて考察します。
ヒトゲノムの構成と構造
ゲノムは、生きた生物で複製されるDNAの総補体と定義されます。自由生活性生物のゲノムの配列決定は1995年に細菌で始まり、酵母(1996年)、線虫(1998年)、ショウジョウバエ(2000年)など、より大きく複雑な生物へと進みました。2001年には、ヒトゲノムの最初のドラフトが完成し、重要な節目を迎えました。現在、何千ものゲノムが配列決定されています。2011年には、ゲノムシーケンシングからのデータがトランスレーショナル医療で使用されることを可能にする完全な技術インフラを提供することを目標に、Genome in a Bottle Consortiumが設立されました。
これらのデータが利用可能になるにつれて、4つの興味深いパターンが現れました。
- 遺伝子の数が予想よりも少ないこと。 ヒトゲノムドラフト完成前の推定では16万個もの遺伝子があると考えられていました。しかし、GENCODEアノテーション(ENCODEプロジェクトの公式アノテーション)の最新版では、60,155個の遺伝子があり、そのうち19,881個がタンパク質をコードする遺伝子です。現在、ホモ・サピエンスには合計で約2万個のタンパク質をコードする遺伝子があることが明らかになっています。
- 遺伝子数は生物の複雑性の予測因子ではないこと。 例えば、アメーバは単純な単細胞生物ですが、ヒトよりもはるかに多くの遺伝子を持つと予測されています(表1.18-1)。マウスとヒトは、ほぼ同数のタンパク質コード領域を持つことが判明しています。
- ゲノムサイズは生物の複雑性の予測因子ではないこと。 アメーバ(Amoeba dubia)は既知のゲノムの中で最も大きく(670ギガベース)—ヒトゲノムの200倍以上です。ヒトとチンパンジーのゲノムは本質的に同じサイズですが、大脳皮質の発達に関して明白で重要な違いがあります。
- 真核生物ゲノムのわずかな割合しかタンパク質をコードしていないこと。 ヒトゲノムのわずか2%しかタンパク質をコードしていません。大部分はイントロンと反復配列で構成されており、その一部は明らかに機能的ですが、その多くは依然として謎に包まれています。タンパク質をコードする遺伝子以外にも、タンパク質をコードしない**非コードRNA(ncRNAs、下記参照)**と呼ばれる、活発に転写されるかなりの数の遺伝子が存在し、これらは時にゲノムの「ダークマター」と呼ばれます。この「ダークマター」の一部は機能的であることが知られていますが、ncRNAの多くが持つ機能的意義はまだ解明されていません。
表1.18-1.
異なる生物間における遺伝子数とゲノムサイズ
| 生物名 | 遺伝子数 | ゲノムサイズ (塩基対) | 染色体数 |
|---|---|---|---|
| アメーバ | 50,000 | 670,000,000,000 | 0 |
| 植物 (シダ) | 37,500 | 100,000,000,000 | 90 |
| ヒト | 25,000 | 3,000,000,000 | 46 |
| チンパンジー | 25,000 | 3,000,000,000 | 48 |
| マウス | 25,000 | 2,500,000,000 | 40 |
| ミツバチ | 15,000 | 300,000,000 | 32 |
| ショウジョウバエ | 14,000 | 130,000,000 | 10 |
| 線虫 | 19,000 | 97,000,000 | 12 |
| 菌類 | 6,000 | 13,000,000 | 32 |
| 細菌 | 3,000 | 5,000,000 | 1 |
| マイコプラズマ・ゲニタリウム | 500 | 580,000 | 1 |
| DNAウイルス | 450 | 50,000 | 1 |
| RNAウイルス | 20 | 10,000 | 1 |
| ウイロイド | 13 | 500 | 1 |
Google スプレッドシートにエクスポート
ゲノムサイズでソートされています。すべての数値はおおよそです。
これらの観察結果は累積的に、精神医学にとって中心的な問いを投げかけています。つまり、ヒトのゲノムが進化的な観点からそれほど特徴的でないとすれば、どのようにしてヒトCNSのユニークな性質を生み出したのか? 最近の進展は、この重要な問いに光を当て始めています。
DNAと染色体
DNAは、アデノシン(A)、シトシン(C)、グアニン(G)、チミン(T)という4種類の核酸、別名ヌクレオチドまたは塩基から構成されています。合計で、ヒトゲノムDNAは約30億個のヌクレオチドから構成されており、この完全な補体は、核を持つ体内のすべての細胞に見られます。核内では、ゲノムは46本のDNA鎖として組織化されており、複数のタンパク質と複合体を形成して染色体を形成します(23本は母親から、23本は父親から受け継がれます)。細胞の核内では、DNA鎖はヒストンタンパク質と結合し、DNAがヒストン-DNA複合体であるヌクレオソームにしっかりと巻きつき、さらにクロマチンと呼ばれる超構造に組織化されることを可能にします。
光学顕微鏡で染色体が最初に観察されて以来、クロマチンは、おなじみの明暗の縞模様に対応する2つのタイプに分けられてきました。すなわち、ユークロマチン(明るく、密度の低い物質)とヘテロクロマチン(暗く、密度の高い物質)です。ある意味では、クロマチンは単にDNAが巻きつく巻き取り器として機能し、ゲノム全体が核内に収まるようにしています。この特性により、細胞は小さく留まることができ、多細胞生物の進化に影響を与えました。しかし、それはゲノムの機能を協調させる上で重要な役割も果たします。DNAが組織化されているヒストンタンパク質のコアは、アセチル化、リン酸化、メチル化などの化学反応によって変化する可能性があります。これらの化学修飾因子の追加または除去は、核内のDNA領域のコンフォメーションを決定することができ、このプロセスは遺伝子発現の調節を協調させるのに役立ちます。エピジェネティクスという研究分野は、クロマチンやDNAメチル化を修飾することによって遺伝子発現が変化するが、DNA配列自体は変化しない方法を研究するために出現しました(本章のエピジェネティクスのセクションを参照)。最後に、クロマチンの組織構造は、染色体の複製に必要なDNAの移動や、細胞分裂(有糸分裂)中にガイドするのに役立つ足場を形成します。
ヘテロクロマチンの濃い染色性は、ヌクレオソームが非常に密にパッキングされているためであり、DNAが細胞の機械にほとんどアクセスできない状態になっています。一方、ユークロマチンの明るい色は、より開いた、ほどけた、そして通常は活性な状態を反映しています。ユークロマチン領域は他の点でも異なっていることが判明しました。それらははるかに多くの遺伝子を持ち、反復配列が少ないです。しかし、ゲノムのヘテロクロマチン領域も活性化することができ、一部のヘテロクロマチン領域は、必要に応じてのみ活性化されるように、相互に修飾されます。これらのプロセスについては、本章の最後にエピジェネティックメカニズムを考察する際に、後でさらに詳しく議論します。
遺伝子、RNA、タンパク質
グレゴール・メンデルは1866年に、彼が「因子」と呼んだ遺伝単位を特定し、エンドウ豆の実験を通してその遺伝の原則を精緻化することで、初めて遺伝子を発見しました。しかし、彼は遺伝子が何から構成されているのかについて物理的またはメカニズム的な理解を持っていませんでした。遺伝子の最初の医学的応用は1902年、アーチボルド・ガロッドが、空気に触れると尿が黒くなることを特徴とする稀な疾患であるアルカプトン尿症がメンデルの遺伝法則に従うことを発見したときに実現しました。彼は、この疾患の伝達が常染色体劣性パターンに従うことを特定しました。しかし、遺伝子が世代から世代へと伝えられ、疾患の責任を担うことは明らかでしたが、正確にどのように、またどの物質で伝えられるのかはまだ不明でした。
1910年、トーマス・モーガンはショウジョウバエの研究を通して、遺伝子が染色体上の離散的な単位であることを実証し、交叉(Fig. 1.18-2)と呼ばれるプロセスで染色体が物質を交換することによる遺伝的連鎖のアイデアを初めて提唱しました。
1941年、エドワード・テイタムとジョージ・ビードルによるパンカビ(Neurospora crassa)の実験は、遺伝子が変異すると特定の酵素が機能しなくなることをさらに示しました。これにより、特定の遺伝子が特定のタンパク質を作るという広く受け入れられた概念、一般に「1遺伝子1酵素」仮説と呼ばれるものが生まれました。
これらの目覚ましい発見にもかかわらず、遺伝情報の伝達のための物理的基盤は依然として議論の的でした。20世紀初頭には、20種類の異なるアミノ酸から構成されることが知られていたため、タンパク質が有利であると考えられていました。DNAはわずか4種類の異なるヌクレオチドから構成されるため、生命を維持するために必要な複雑な指示を作るために必要な多様性を可能にするのはタンパク質である可能性が最も高いと考えられていました。しかし、1944年、オズワルド・エイブリーによって、DNAが細菌において遺伝可能な形質転換を引き起こすことができるが、タンパク質はできないことを実証することにより、遺伝子がDNAで構成されていることが示されました。

図1.18-2. 交叉(Crossing Over)
この図は、染色体(円)に沿って配置された遺伝子が、減数分裂中に異なる一連の遺伝子(黒い円に突然白い円が加わっている)と情報を交換する(交叉)模式図です。
遺伝子が染色体上で離れているほど、それらが交叉する頻度は高くなります。この現象は**組換え(recombination)**として知られています。
そしてついに、1953年、ワトソンとクリックはDNAの化学構造を発表し、それが2つの糖リン酸骨格が4つのヌクレオチドを支える二重らせんであることを示しました。これらのヌクレオチドは、一方の鎖の「A」が他方の鎖の「T」と、一方の「C」が他方の「G」と特定の対を形成することが認識されました。
コーディング遺伝子
過去数十年の進歩により、大きく分けて2種類の遺伝子があることが認識されるようになりました。
- 直接タンパク質の生産につながるもの(コーディング遺伝子)
- そうでないもの(非コーディング遺伝子)
コーディング遺伝子は、酵素(RNAポリメラーゼ)によって一時的なmRNAに転写され、それが分子生物学のセントラルドグマとして知られるものに従ってタンパク質(ペプチドで構成)に翻訳されるDNAのストレッチです。すなわち、DNA → RNA → タンパク質(図1.18-3)。
1960年代には、コーディングDNAがコドンと呼ばれる3文字のセグメントで「読まれる」ことが発見されました(図1.18-3を参照)。各コドンは、ペプチドまたはタンパク質中のアミノ酸となります。しかし、より単純な原核生物とは異なり、ヒトのコーディング遺伝子は、最初に編集が必要な配列として転写されます。コーディング遺伝子には、コドンとして読まれるセグメント(エキソンと呼ばれる)と、転写されるがタンパク質には翻訳されない非コード配列(イントロンと呼ばれる)の両方があります。非コード配列は、遺伝子の3つの領域に存在し得ます。すなわち、遺伝子の前(転写の順序に対して上流)、遺伝子の内部、および遺伝子の後(下流)です。遺伝子の上流の配列は5′ UTRと呼ばれます。コーディング領域の下流の非コード領域は3′ UTRです。成熟mRNAには、転写機構にポリAテールとして知られる長いアデノシンの鎖をmRNAの3’末端に挿入するように指示するポリアデニル化シグナル(AAUAAなど)が含まれています。単一の遺伝子には、その3′ UTRに複数のポリアデニル化シグナルが連続して含まれることがありますが、どれが使用されるかを決定するメカニズムはまだ確立されていません。複数のポリアデニル化シグナルは、単一の遺伝子から可変的な3′ UTR長を持つ転写産物が生成されることを可能にします。より長い3′ UTRを持つ転写産物は脳で比較的普及しており、RNA制御の微調整を可能にするより多くのmiRNA結合部位を生じさせます。さらに、より長い3′ UTRは、タンパク質を特定の細胞内位置にシャトルするタンパク質結合モチーフを宿すことが知られています。BDNF遺伝子は、BDNFを樹状突起に輸送可能にする結合モチーフを持つ長い3′ UTRを持つ形態を有することが知られています。したがって、コーディング遺伝子から産生される新生mRNA転写産物は、適切な細胞内位置でタンパク質産物を生産するために必要な首尾一貫した一連の指示につながるように、まず処理される必要があります。遺伝子スプライシングと呼ばれるプロセスによるイントロンの除去は、遺伝子の機能を決定する上で重要なステップであり、ゲノムの各部分の機能を考察する際にさらに詳しく説明されます。
遺伝子転写産物は3塩基コドンで読まれ、DNAは4種類のヌクレオチドで構成されているため、64通りの可能なコドン(4 x 4 x 4)があります。ほとんどの生物では、タンパク質に翻訳されるmRNAは、開始コドン(AUG)と呼ばれる3塩基配列を持ち、コドンの読み取りまたは翻訳がどこから始まるかを区別します。3つの終止コドン(UAA, UAG, UGA)は、伸びるポリペプチド鎖の最後のアミノ酸を合図します。開始コドンと終止コドンは遺伝子を読むために必要な句読点であり、他の60個のコドンは、ペプチドまたはタンパク質に含めるアミノ酸を決定します。細胞はタンパク質を作るために20種類の異なるアミノ酸しか使用しないため、異なるコドンが同じアミノ酸をコードできるという遺伝コードに冗長性が存在します(図1.18-4)。このコドン使用における冗長性は、特定の機能を持つ可能性があります。コドン使用バイアスと呼ばれるものがあり、翻訳が特定のコドンを他のコドンよりも優先します。コドン翻訳効率の違いは、それらのコドンに対応するtRNAの濃度と関連しており、それらの間に複雑な相互作用があることを示唆しています。このような複雑な進化的変化は、特にニューロンのように非常に幅広い発現遺伝子のレパートリーを持つ組織において、翻訳プロセスを微調整する可能性があります。
処理されたRNAは、細胞内をリボソームと呼ばれるオルガネラに運ばれ、タンパク質鎖に変換されます。このタンパク質鎖は、その後、ますます複雑な組織レベルに折り畳まれていきます(表1.18-2)。
ゲノムによって作られるすべてのタンパク質の総体はプロテオームと呼ばれます。多くのタンパク質は、より大きなタンパク質複合体(転写を行う大きな酵素であるRNAポリメラーゼIIを含む)のサブユニットとして機能し、多くのタンパク質を一度に研究することはプロテオミクスとして知られており、これも本章で後ほど詳しく説明されます。
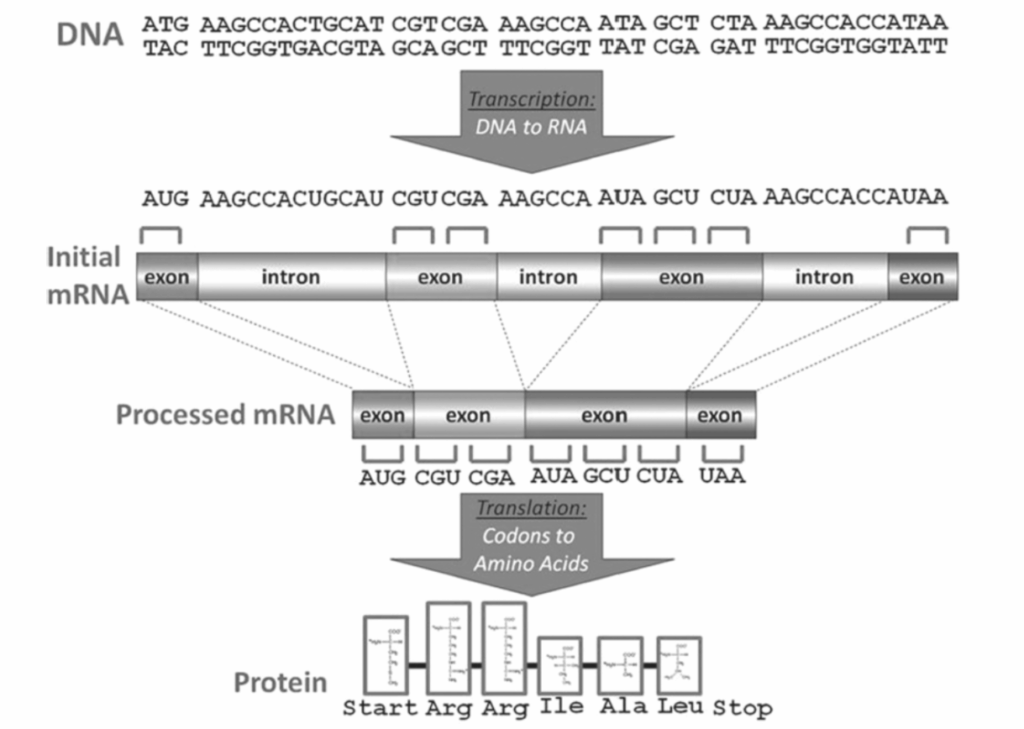
図1.18-3. 分子生物学のセントラルドグマ
転写中に、DNAのチミン(T)は異なる核酸(ウラシルを示すU)で表され、コドンが細胞の機構によって理解されるようになります。
イントロンは最終的なメッセンジャーRNA(mRNA)から除去され、コドンの翻訳を通じてタンパク質になる準備ができた産物が残ります。
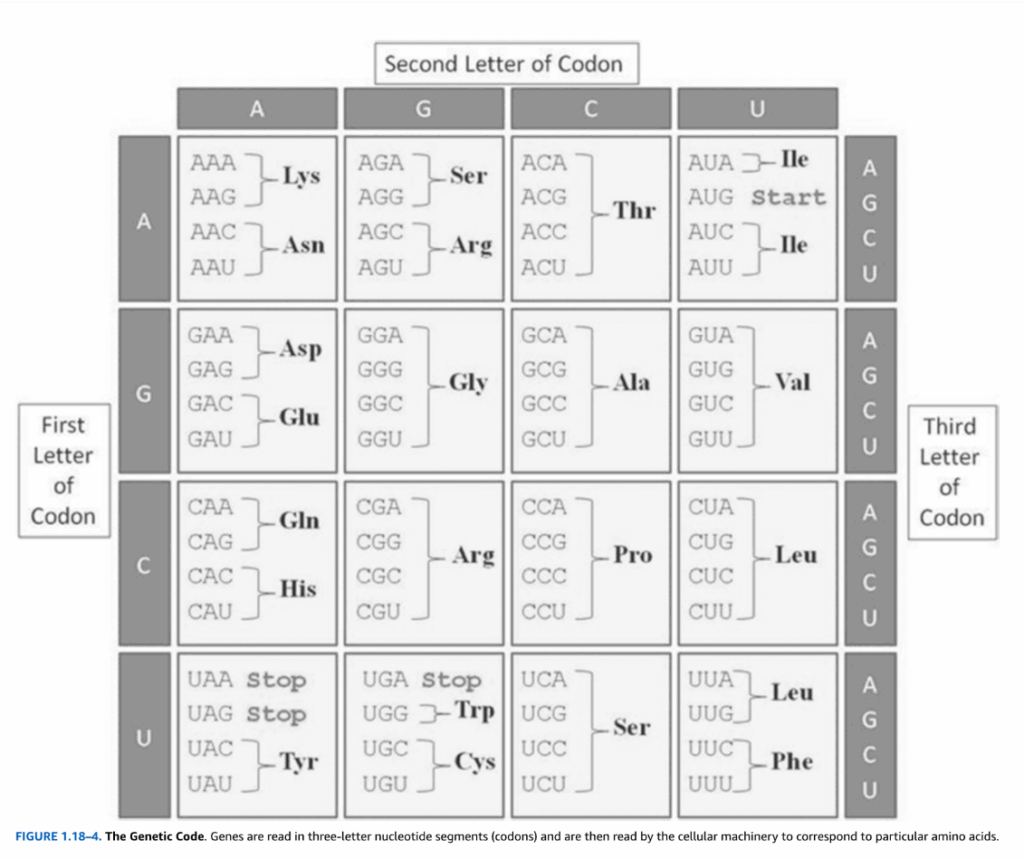
図1.18-4. 遺伝コード
遺伝子は、3文字のヌクレオチドセグメント(コドン)として読まれ、その後、細胞の機械によって特定のアミノ酸に対応するものとして解読されます。
表1.18-2.
タンパク質構造の定義
| 名称 | 定義 |
|---|---|
| 一次構造 | アミノ酸(ペプチド)の鎖で、ポリペプチドを形成する |
| 二次構造 | アミノ酸が鎖内で水素結合によって結合している |
| 三次構造 | ポリペプチドが全体としてシートやヘリックスを形成し、3D構造をとる |
| 四次構造 | 複数のポリペプチド鎖が結合して、高分子構造を形成する |
Google スプレッドシートにエクスポート
コーディング遺伝子は、遺伝情報がどのように保存され処理されるかという初期の概念に最も密接に適合していますが、時間の経過とともに、これらがゲノム全体のわずか約2%に過ぎないことが明らかになりました。さらに、DNA内に含まれる別の指示のセットが、タンパク質の生産には続かない転写を支配しています。これまでにヒトゲノム内で3万以上の非コーディング遺伝子がカタログ化されています。これらはRNAで構成されており(したがって、非コーディングRNA、またはncRNAとも呼ばれます)。表1.18-4にいくつかのRNAの種類がリストされています。一部は遺伝子転写およびタンパク質翻訳の正常な細胞プロセスを調節することに関与していますが、他のものは最近発見されたばかりであり、その機能はまだ十分に理解されていません。さらに、一部のncRNAは単独で機能し、反応を触媒したり、細胞内で自律的に作用したりすることができます。
最もよく特徴づけられているncRNAの一部はsiRNAと呼ばれています。これらはDNAにコードされた転写産物であり、ゲノムからの別の転写配列と相補的(またはアンチセンス)です。siRNAが相補的なmRNA配列に結合すると、細胞はその転写産物を分解し、したがってDNAまたはmRNA配列を変化させることなく、mRNAを産生した遺伝子を機能的にサイレンシングします。同様に、非コーディング遺伝子の異なるサブセットである**マイクロRNA(miRNA)**は、短鎖(18〜22 bp)の配列であり、ほとんどの場合、転写産物の3′ UTRに結合し、最も一般的には結果として生じるタンパク質のレベルを減少させます。siRNAとmiRNAは両方とも、転写後に遺伝子発現を調節するために機能します。したがって、DNAからタンパク質へのプロセスにおいて遺伝子発現が変化する複数の極めて重要な方法は、ゲノムの機能を理解しようとする科学者にとって非常に重要です。このトピックは本章の第2節で扱われます。ncRNAが広く存在し、遺伝子発現の調節において重要な役割を果たすという認識は、それらが精神疾患や他の複雑な疾患のリスクを付与する可能性が強いことを強く示唆しています。さらに、siRNAが特定の遺伝子の発現喪失の結果を細胞レベルまたはモデル生物レベルで評価するのに役立つことがすぐに認識されました。その結果、siRNAは迅速に不可欠な研究ツールとなりました。
表1.18-3.
GENCODE 2010-2020における、コード能力とバイオタイプによるアノテーション遺伝子の分布
| GENCODE v37 (2020年8月) | GENCODE v21 (2014年6月) | GENCODE v10 (2011年7月) | GENCODE v7 (2010年12月) | |
|---|---|---|---|---|
| 総遺伝子数 | 60,651 | 60,155 | 52,376 | 51,082 |
| タンパク質コード遺伝子 | 19,951 | 19,881 | 20,007 | 20,687 |
| 長鎖非コーディングRNA遺伝子 | 17,948 | 15,877 | 10,840 | 9,640 |
| 短鎖非コーディングRNA遺伝子 | 7,569 | 9,534 | 8,801 | 8,801 |
| 偽遺伝子 | 14,773 | 14,467 | 12,358 | 11,580 |
Google スプレッドシートにエクスポート
ncRNAの他の2つの重要なクラスは、偽遺伝子とLINCです。偽遺伝子は脳トランスクリプトームに遍在し、いくつかのタイプの転写産物を含みます。コード能力を失った転写活性な遺伝子であるユニット偽遺伝子(GULO-ビタミンC産生酵素)、コード能力を失った重複遺伝子、および逆転写された遺伝子(処理された偽遺伝子)です。それらの数が後続のアノテーションごとに増え続けている理由は、それらが機能性遺伝子と実質的な配列を共有しているためであり、これがそれらの特定を極めて困難にしています。バイオインフォマティシャンは、偽遺伝子を機能性遺伝子から区別できるようにするための高度に洗練されたアルゴリズムを開発してきました。「スポンジ仮説」は、偽遺伝子が限られたmiRNAのプールを機能性遺伝子と競合する可能性があることを示唆しています。もしそうであれば、偽遺伝子は、本来の機能を持たなくても、全体的な遺伝子調節に影響を与える可能性があります。
LINCも神経機能に影響を与えるようです。ほとんどのLINCは、様々な方法で転写(H19、XIST、AIR、HOTAIR)を変化させます(すなわち、転写への直接的な影響、エピジェネティックシグネチャの変更など)。他のものは翻訳レベルで機能を発揮します(GAS5)。
かなりの量のncDNAが機能的なncRNAに転写されますが(表1.18-4を参照)、ゲノムのどれだけが活発に処理されているのか、またこの転写が生物学的機能にどれだけ直接寄与しているのかは明確ではありませんでした。この疑問に答えるため、国立ヒトゲノム研究所(NHGRI)は2003年に、ゲノム内のすべての機能要素を特定するENCODE(DNA要素の百科事典)プロジェクトを立ち上げました。ENCODEプロジェクトの第2段階までには、ヒトゲノムの75%が転写活性があるように見えました。しかし、ゲノムの70%は極めて低い発現レベルを示しており、機能的意義に関する疑問を提起しています。また、ゲノムの11%は、1つ以上の細胞型における転写因子結合領域のモチーフまたは高解像度DNaseフットプリントに関連していました。プロモーター関連ヒストン修飾またはエンハンサーはゲノムの約20%をマークしており、ゲノムの3分の1もの部分が転写伸長に関連する修飾によってマークされていました。これらを総合すると、ゲノムの大部分が転写されているか、または調節要素を宿しているように見えます(図1.18-5)。
これらの観察結果は、遺伝物質を分類する能力を限界にまで押しやりました。コーディング遺伝子と非コーディング遺伝子の間には明確な区別がありますが、大量の転写活性を持つDNAは、現在の機能カテゴリに適合しないRNAを生成します。明確にするために、ncRNAという用語は、siRNAやmiRNAなど、その一般的な構造がよく特徴づけられている非コーディング遺伝子を記述するために使用されます。追加の転写された物質は、**機能不明の転写産物(TUFs)または転写活性領域(TARs)**と呼ばれます。
TUFsとTARsは、以下を表している可能性があります。
- 以前の実験や遺伝子発見アルゴリズムで見逃された新しいタンパク質コーディング遺伝子
- 新しい非コーディング遺伝子
- 他の遺伝子を調節する可能性のある新規なアンチセンス転写単位(siRNAおよびmiRNA)
- 最終的なmRNA産物にイントロン領域を含む既知の遺伝子の代替アイソフォーム
- 長くすべきか短くすべきか、2つの遺伝子に分割すべきか、1つの大きな遺伝子に統合すべきか誤ってアノテーションされた遺伝子
- 異常な転写または偶発的な転写読み抜けを表す生物学的アーティファクト
- 少量のゲノムDNA汚染による実験的アーティファクト
表1.18-4.
RNAの種類
| 名称 | 略語 | 発見年 | 機能 |
|---|---|---|---|
| メッセンジャー | mRNA | 1956 | DNAからの転写されたメッセージを運ぶ |
| リボソーム | rRNA | 1958 | タンパク質合成機構であるリボソームの構成要素 |
| 転移 | tRNA | 1962 | 翻訳においてアミノ酸をリボソームに運ぶ役割をする |
| 小核 | snRNA | 1977 | 未成熟mRNAのスプライシングを助け、最終形態に到達させる |
| 小核小体 | snoRNA | 1986 | rRNAおよび他のRNAの化学修飾を直接的に指示する |
| マイクロ | miRNA | 1993 | 遺伝子発現を調節する一本鎖RNA |
| 短鎖干渉 | siRNA | 1999 | 転写を妨害する短鎖二本鎖RNA |
| Piwi作用性 | piRNA | 2006 | 生殖細胞系に作用するRNAで、寄生性遺伝要素を阻止する |
| ウイルス由来短鎖干渉 | viRNAs | 2011 | ウイルスサイレンシングの世代間遺伝を媒介するウイルス由来の短鎖干渉RNA |
Google スプレッドシートにエクスポート
将来の実験によってこれらのTARの役割が明確になる必要がありますが、いくつかの結論はすでに可能です。例えば、ユークロマチンゲノムの大部分(93%)は何らかの能力で機能しています(転写されているか、別の配列を調節しているかのいずれかです)。また、遺伝子調節はゲノム全体で対称的であり、調節構造の上流または下流への配置に偏りはありません。これは、遺伝子の開始コドンより5’または上流の領域がその転写産物の調節を最も変調させる可能性が高いという以前の概念とは対照的です。最後に、驚くべきことに、ゲノムのこれらの調節領域または活性領域の多くは、種間で高度に保存されていません。これらのデータを総合すると、脳の発達や精神疾患に寄与する遺伝子や遺伝的メカニズムの探索は、コーディング配列の範囲をはるかに超えて拡張する必要があることが示されています。
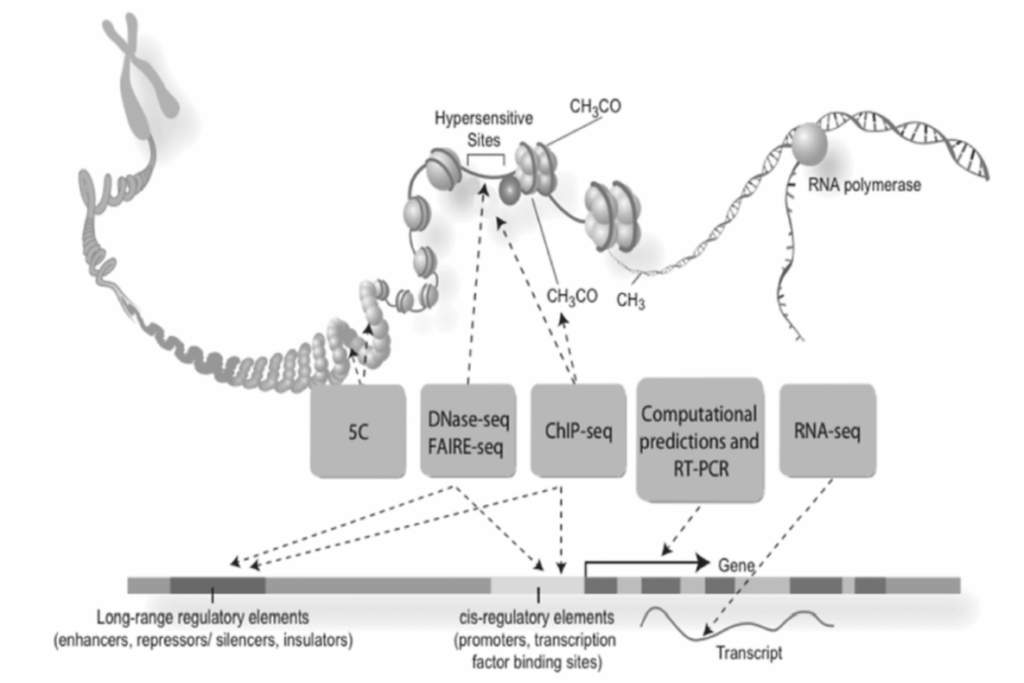
図1.18-5. ENCODEプロジェクトのまとめ
この図は、ENCODEプロジェクトの概要を示しています。
TARs(転写活性領域)の発見とアノテーションは、多様な細胞株からRNAをシーケンシングし、その後、比較ゲノミクス、統合バイオインフォマティクス手法、およびヒトによるキュレーション(精査)を行うことによって実施されました。
調節要素の調査には、DNA過敏性アッセイ、DNAメチル化アッセイ、および修飾ヒストンや転写因子を含むDNAと相互作用するタンパク質の**クロマチン免疫沈降(ChIP)**を行い、その後シーケンシング(ChIP-Seq)を行うことが含まれました。
繰り返しDNA
ヒトゲノムの配列決定と、ごく一部のみがコーディングDNAまたはncRNAの形で存在するという認識により、ゲノムの残りの部分への関心が高まりました。この物質の大部分は、繰り返し要素の形で存在しています。
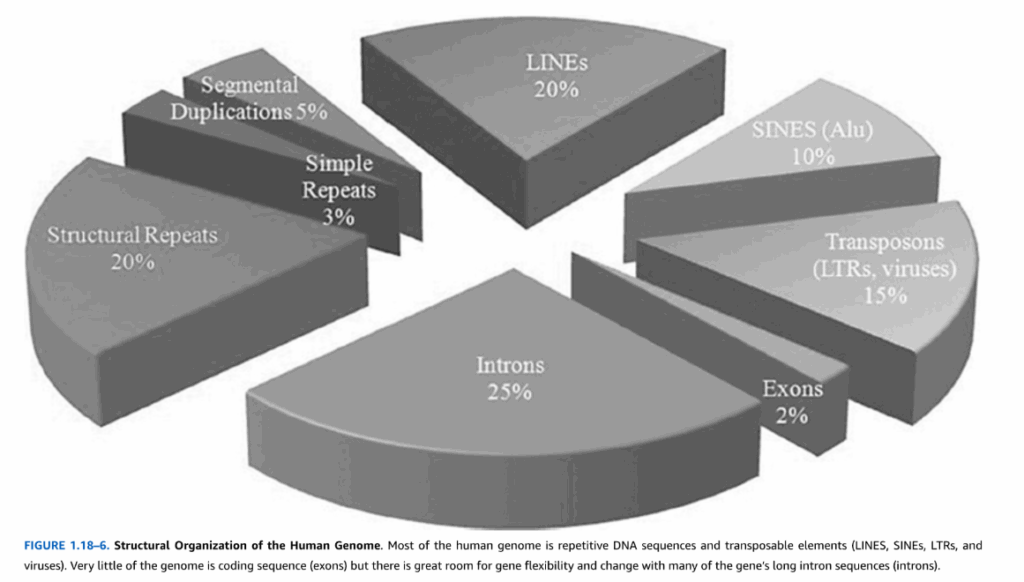
図1.18-6. ヒトゲノムの構造的構成
ヒトゲノムの構造的構成を図1.18-6に示します。ヒトゲノムのほとんどは、反復DNA配列と**転移因子(LINES、SINEs、LTRs、およびウイルス)**で構成されています。
ゲノムの**コーディング配列(エキソン)はごくわずかですが、遺伝子の長いイントロン配列(イントロン)**には、遺伝子の柔軟性と変化の大きな余地があります。
ヒトゲノムの構造的構成は図1.18-6に示されています。ゲノムのほぼ3分の1は、反復要素で構成されています。ヒトゲノムのいくつかの反復セクションは、染色体の末端または中央で構造的役割を果たしています。しかし、単純な反復配列(SSRs)やセグメンタルデュプリケーション(SegDups)を含む、多くの種類の反復の機能は不明です。
SSRsは、ゲノム内でタンデムに繰り返される短い配列(2〜6 bp)です。ハンチンチンタンパク質をコードする責任のあるHGG遺伝子のコーディング部分にある短い三ヌクレオチド反復(CAG)は、最終的に致命的な進行性神経変性疾患であるハンチントン病の原因であることが知られています。健康な個人ではHGG遺伝子に10〜35個の繰り返しがありますが、罹患した個人では最大120個の繰り返しを持つことがあります。疾患の発症と重症度は、繰り返しの数と非常に強く相関しています。これは、症状発現前に簡単な検査で診断できる、明確に定義された遺伝的病因を持つ数少ない神経精神疾患の1つです。ハンチントン病の親を持つ子供は、この障害を発症するリスクが50%です。したがって、症状発現前の個人において疾患を診断できる能力は、神経精神医学の臨床実践における遺伝学の役割に関連する重要な倫理的問題を提起します。
SegDupsは、ゲノム全体に少なくとも2回出現する1,000 bp以上の配列です。興味深いことに、これらのSegDupsの一部には、遺伝子全体の余分なコピーが含まれています。
ゲノム内で最も遍在する反復遺伝要素は転移因子(TEsまたはトランスポゾン)であり、ゲノム内のある場所から別の場所にジャンプすることができます。このプロセスは、人間にとって有用な新しい形態の遺伝子につながる可能性がありますが、必須遺伝子を破壊する危険性も伴います。TEはいくつかのサブグループに分けられます。長鎖散在配列(LINEs)、短鎖散在配列(SINEs)、および短い300 bpのAlu要素(SINEの一種とみなされます)。Alu要素はイントロンの75%に存在し、ゲノム全体の10%を占める一方、6,000 bp(6 kb)のLINEsはヒトゲノムの20%を占め、何千もの遺伝子(5′ UTR、エキソン、3′ UTR、イントロン)で見られます。ヒトゲノムのほぼすべての遺伝子は、少なくとも1つのTEを持っています。
Alu要素はTEsであると同時にレトロトランスポゾンでもあり、7SL RNA由来の霊長類特異的リピートファミリーのようなレトロウイルスの方法で逆転写を介してDNAを変化させます。霊長類系統の進化全体でのそれらの分布は、ヒトゲノムに最大110万コピーもの存在をもたらしました。Alu要素の挿入は、いくつかの主要な波で起こり、古いAluJサブファミリー(AluJoとAluJbを含む;約8100万年前)、中年期のAluSサブファミリー(AluS、AluSx、AluSg、AluSq、AluSp、AluSc、AluSbを含む;約4800万〜1900万年前)、そして若くてまだ活動的なAluY波(AluY、AluYa5、AluYa8、AluYb8を含む;約600万年前に始まり現在も継続中)を派生させました(M7)。ゲノムに遍在しているにもかかわらず、Aluリピート(他のリピートまたは低複雑性配列と同様に)は、現在のショートリードシーケンシングを使用して正確にマッピングすることが極めて困難です。この事実と、霊長類から得られた限られたRNA配列データが、これらの要素の適切なマッピングの欠如を説明しています。Alu要素の本体は長さ約300 bpで、短いAリッチ領域によって分離された2つの分岐したダイマーから形成されています。このような構造により、それらは完全に二本鎖構造に折り畳むことができ、したがってRNA編集と呼ばれるプロセスの基質となります(下記で説明)。新しい発見は、Alu要素が培養ニューロンだけでなく、生体内ニューロンでも活発に転移していることを示しています。神経ゲノムにおけるこれらの変化は、独自の遺伝的、トランスクリプトーム的、プロテオーム的構成を持つニューロンを生成し、脳の複雑性を増加させる可能性があります。
ヒトとチンパンジーとの共通祖先からの分岐以来、約98,000のウイルスがヒトゲノムに侵入してきました。これら内在性レトロウイルスと呼ばれるウイルスは、ヒトゲノムの8%を占め、RNAからDNAへと自身を逆転写する長末端反復配列(LTRs)、DNA TEs、および休眠状態にありもはや複製できない一部のウイルス(ヒトゲノムの約4%)で構成されています。これらの配列の一部は、本質的に死んだゲノムであり、ヒトDNAに取り込まれ、現在ゲノムと共に複製されていますが、他のものはレトロトランスポジションによって依然として活発にヒトを感染させています。
ヒトゲノムに存在する膨大な量の反復性非コーディングDNAは、ヒトゲノム全体の98%を占めます。これらの領域は機能しないと考えられ、「ジャンクDNA」という誤称を得ていました。しかし、非コーディングDNAに帰せられる多くの機能に鑑み、ゲノムにおけるこの「ダークマター」の機能はまだ発見されていないというのが一般的な見方です。たとえ特定の配列の機能が特定されていなくても、それが近くの、あるいは驚くほど遠く離れた他の遺伝子の機能にとって依然として重要である可能性があります。たとえある配列が、ゲノムの何千もの場所に存在する反復性TEであっても、単一のTEの場所が、その要素を持つ特定の遺伝子の正しい活動に必要となる場合があります。わずか10年前とは完全に逆転して、現在ではゲノムのすべての配列が機能的であると推定され、精神医学におけるゲノム生物学のより良い理解への扉を開き、徹底的な検討を必要とするはるかに大きなゲノムを明らかにしています。
ミトコンドリアゲノム
ヒトを含むすべての真核生物は、2つの完全に機能する生きたゲノム、すなわちその種に固有のDNAと、ミトコンドリア(動物の場合)または葉緑体(植物の場合)のゲノムを持っています。細胞内共生説は、地球上の生命が進化し始めた初期の時代(15億〜20億年前)に、多細胞生命体が呼吸細菌と融合し、よりアクセスしやすい形で細胞エネルギー(ATP)を生産できるようになったと示唆しています。この関係は、細菌が捕食を免れることができ、原真核生物が新しいエネルギー源を得たため、両者にとって相互に有益であることが証明されました。この説は、ミトコンドリアが独自のゲノムと独立した複製サイクルを持ち、ミトコンドリアの最も近い現存する近縁種(リケッチア細菌)が真核細胞の寄生虫であるという事実によって裏付けられています。
ヒトゲノムとミトコンドリアゲノムは、現在完全に絡み合った共存関係にあります。ミトコンドリア由来の約300個の遺伝子が、ヒトの23本の染色体上に存在します。これらはミトコンドリア起源の核DNA(NUMTs)と呼ばれ、ミトコンドリアの機能をサポートするために核ゲノムによって産生されます。さらに、これらのNUMTsのうち27個はチンパンジーや他のゲノムには現れず、したがって過去400万〜600万年の間にヒトゲノムに組み込まれてきました。それらのほとんど(27個中23個)は、既知または予測されるヒト遺伝子内に存在しており、これらの2つのゲノム間の共生が有意に遺伝子中心であることを示しています。これはおそらく、転移因子に対するより強力な保護、および/または活性酸素種(ROS)による損傷からミトコンドリアDNA(mtDNA)を保護することを確実にしていると考えられます。
さらに、一部の神経筋疾患や代謝欠陥は、mtDNAまたはNUMTsに特異的な変異やエラーから生じます(表1.18-5)。これは、精神疾患のリスクに影響を与える上でミトコンドリアゲノムの重要性を強調しています。最後に、最近の研究は、ミトコンドリアがヒトの生理機能、特にストレスに対する身体の反応にどれほど不可欠であるかを示しています。宇宙飛行士のプロファイリングに対するマルチオミクスおよびシステム生物学のアプローチは、宇宙飛行の一貫した表現型として、ミトコンドリアプロセスが有意に濃縮されていることを明らかにしました。
エクソソーム
エクソソームは、細胞が分泌するDNA、RNA、タンパク質を含む細胞外小胞の一種です。エクソソームの生理学的目的は、大部分が不明であり、さらなる調査が必要です。一部の理論家は、エクソソームが過剰または不要な分子を除去することによって細胞のホメオスタシス維持に役割を果たすと仮定していますが、他の理論家は、エクソソームが複雑な細胞間および組織間コミュニケーションを調節する上で不可欠であると信じています。
実際、研究により、エクソソームは心血管疾患、ウイルス病原性、癌の進行、炎症性疾患、および神経変性を含むCNS関連疾患と関連していることが示されています。これらの知見は、エクソソームの分子メカニズムと固有の特性が、これらの疾患の治療的制御または診断評価において潜在的な有用性を持つ可能性があることを示唆しています。また、エクソソームは、治療薬や免疫調節剤を特定の標的に送達するように設計またはプログラムすることができます。さらに、それらはすべての生体液中で報告されており、液体生検によって容易にアクセスできることが判明しているため、臨床医にとって潜在的に有用な診断および予後マーカーとなります。
エクソソームは、脳内の誤って折り畳まれたタンパク質の凝集を促進および制限の両方を行うことが示されており、神経変性や他の神経疾患の病因と治療の両方において潜在的な役割を果たす可能性があります。薬物や毒素が脳に到達するのを防ぐ選択的フィルターとして機能する血管網である血液脳関門を効率的に通過できるというそれらのユニークな固有の特性は、エクソソームを研究者や臨床医にとって重要な治療標的として注目させています。
表1.18-5.
ミトコンドリアによってコードされる遺伝子に起因するヒト疾患
| 疾患名 | 影響を受ける遺伝子 |
|---|---|
| NARP(神経障害、運動失調、色素性網膜炎) | ATPase6 |
| MELAS(ミトコンドリア脳筋症、乳酸アシドーシス、脳卒中様発作) | tRNA (Leu), Cytochrome C |
| MERRF(ミオクローヌスてんかん;不整赤色線維) | tRNA (Lys), tRNA (Ser) |
| LHON(レーバー遺伝性視神経症) | MTND4 |
| フリードライヒ運動失調症 | FRDA |
| リー症候群 | ATPase6, MTND5 |
Google スプレッドシートにエクスポート
ヒトの遺伝的多様性
2001年の参照ヒトの全ゲノムシーケンシングは、個人間および集団間の遺伝的多様性に関する集中的な研究の舞台を整えました。一般的に、互いに関係のない2人の間には、約1,000塩基対に1つの違い、すなわちゲノムの0.1%の違いが存在します。また、誰もがゲノムのすべての部分の2つのコピーを持っているわけではないようです。これらのより大きな可変領域(>1 kb)は、ヘテロクロマチン領域とユークロマチン領域の両方で発生し、ヒトゲノム全配列の5%から20%を占めると推定されています。これらの変異の有病率は、ヒト種内の多様性の総負担に関する考え方を劇的に変えました。精神医学における疾患感受性遺伝子の探索は、精神病理の原因となるか、またはそれに寄与する遺伝的多様性のわずかな割合を探索することであると、より正確に考えることができます。
配列の多様性
個人のDNA配列は、ヌクレオチドレベルでの遺伝コードに関して異なります。このような変化には、あるヌクレオチドの別のヌクレオチドへの置換、または場合によっては1つまたは少数の塩基の挿入または欠失(インデル)が含まれることがあります。さらに、DNA内の短い反復配列は、これらの領域に存在するヌクレオチドの数に関して異なる場合があり、これらの変異は**短鎖タンデムリピート(STRs)および短鎖配列反復(SSRs)**として知られています。
変異を一般的なものとして分類するための閾値は様々です。通常、配列変異が人口の1%未満で見られる場合、それは稀であると見なされますが、一部の著者では5%の閾値を使用しています。特定の集団でこの頻度を超えて見つかる特定の遺伝子変異は、通常多型と呼ばれますが、この用語は、その頻度に関係なく、またそれがコードまたは調節するRNAまたはタンパク質の機能に有害であるかどうかにかかわらず、ゲノム内のいかなる変化にも適用されることがあります。同様に、DNAヌクレオチド配列のいかなる永続的な変化も変異と呼ぶことができますが、この用語は疾患関連の遺伝的変異に最も一般的に適用されます。
集団頻度が1%を超える一般的な単一塩基置換は、しばしば**SNP(一塩基多型)**と呼ばれます。これらは集中的に研究され、遺伝子発見の取り組みにとって不可欠であることが証明されています。SNPは人類の歴史において自発的に発生し、その歴史のある時点で一度だけ起こり、その後時間をかけて集団全体に分布したと考えられています。SNPは一般的に世代から世代へ安定しており、人類が進化するにつれて非常に多数がゲノムに蓄積してきました。現在、3000万の既知のSNPが特定されています。これらの個々のSNP、または単一染色体上のSNPの配置(ハプロタイプと呼ばれる)により、科学者は家族内の遺伝を追跡したり、集団内で疾患のリスクを伴う可能性のある変異を特定したりすることができます。さらに、様々な集団間でのSNPとハプロタイプの分布は、人類の進化に関する重要な洞察を提供してきました。例えば、この種のデータは他の遺伝的および考古学的証拠とともに、人類がアフリカのヒト族から起源したという説に対する強力な証拠を提供してきました。
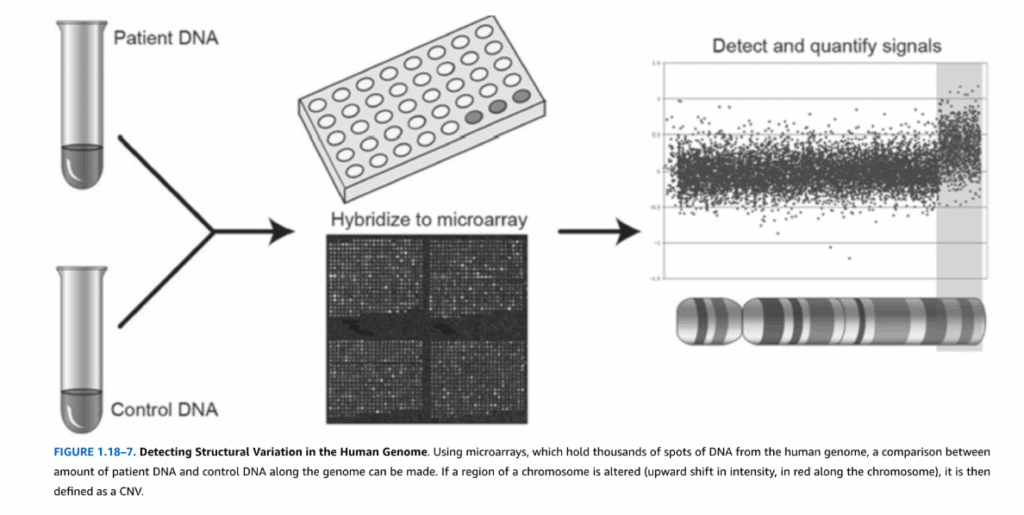
図1.18-7. ヒトゲノムにおける構造的変異の検出
この図は、ヒトゲノムの何千ものDNAスポットを持つマイクロアレイを使用して、患者DNA量と対照DNA量間の比較をゲノム全体で行う方法を示しています。
染色体のある領域が変化している場合(赤色の強度の上方シフト、染色体に沿って)、それは**CNV(コピー数変異)**と定義されます。
構造的変異
バランス型およびアンバランス型転座、大規模な欠失または逆位、および余分な染色体を含む大規模な顕微鏡的変異は何十年も研究されてきましたが、最近になって、ゲノムには膨大な量の準顕微鏡的構造的変異も存在することが明らかになりました。
1990年代後半、いくつかの研究室が、光学顕微鏡の分解能以下の染色体異常を特定するツールを開発しました。染色体異常と癌や精神遅滞を含む様々なヒト疾患との既知の関係を考えると、これまで見えなかった染色体異常を特定する能力が、追加の疾患感受性遺伝子の特定につながると予想されていました。これらの努力の驚くべき結果は、技術の検証に使用された対照サンプル間で、染色体セグメントのコピー数に違いが特定されたことでした。これらの変化が明らかな表現型を持たない個人間で広く存在するという明確な証拠があるため、これらの変化は**CNP(コピー数多型)またはCNV(コピー数変異)**として知られるようになりました。
その後の研究は、これらの初期の結果を広く再現し、より高解像度の技術の開発は、各遺伝子座の予想される2コピーからの逸脱がゲノム全体に一般的であることを示しました。これらの変異は、配列の置き換えや変化(突然変異やSNPのように)ではなく、インデルに似ており、ゲノムの領域が1コピー欠失している(ヘテロ接合性欠失)、両方のコピーが欠失している(ホモ接合性欠失)、または余分なコピーを持っている(増幅)場合です。健康な人間は、数十から数百のCNVを持ち、合計で千ものCNVを持つことがあります。特定されたすべての変異(SNP、CNV、およびより大規模なDNA欠失および/または増幅)の総和を考慮すると、ゲノムの最大20%が個人間で異なることが示されています(図1.18-7)。
遺伝子(および大規模なゲノムセグメント)の重複は、ヒト科系統の進化の過程で多くの進化的変化をもたらしました。三色覚は、約4000万年前に霊長類系統で遺伝子重複の結果として発達しました。このような再配置は、オプシン遺伝子に隣接するAluリピートによって促進され、重複が起こることを可能にしました。チトクロームP450酵素をコードする遺伝子は、ヒトにおいて頻繁にCNVを含んでいます。これらは薬物代謝の重要な酵素であり、そのため、薬理ゲノミクスという新しい分野(下記で説明)で極めて重要です。
CNVの発見は、計り知れない影響を与えました。発達に影響を与える遺伝子を含むヒトゲノムの約10%の遺伝子が、一般的なCNVと重複しており、これはヒトゲノムが以前予想されていたよりもはるかに大きな程度でハプロ不全(2つの半数体コピーのうちの1つの喪失)を許容できることを示唆しています。いくつかのメカニズムがCNV形成につながります。一部のCNVは、ヒトの歴史で一度発生し、その後集団間で分布したと考えられています。他のCNVはより動的であるようです。この場合、ゲノムの領域はCNVにつながるように準備されているかもしれませんが、このプロセスは異なる個体間でランダムに発生し、単一の染色体領域に複数の異なるCNVを引き起こす可能性があります。
CNVとヒト疾患との関係は、集中的な研究対象です。現在、構造的変異に関する遺伝子型と表現型の関係は非常に多様であることが明らかになっています。以前は、精神遅滞の評価を受けている患者で欠失が特定された場合、その欠失が臨床的問題の可能性のある原因であると推定されていました。現在では、そのような患者で遺伝子の喪失が特定された場合、たとえそれが脳の発達に関与していることが知られている遺伝子であっても、もはや因果関係の第一義的な証拠ではありません。現在では、一般的に以下のことが推定されています。
- 一部のCNVには表現型の影響がない。
- 他の種類の変異と同様に、いくつかの一般的なCNVは、複雑な多遺伝子疾患に徐々に寄与することが判明するだろう。
- いくつかのコピー数変化、特に稀なものや**de novo(単一の個人で新しく生成された)**なものは、一部の疾患に対して大きなリスクを伴う可能性がある。
ヒトゲノムにおけるこれらの種類の変異の特性評価が初期段階にあることを考えると、特定の患者または集団においてこれらの選択肢を区別することは依然として困難な課題です。
一般的変異と稀な遺伝的変異
遺伝的変異が配列に基づくものであろうと構造的であろうと、いったんヒトゲノムに導入されると、それは自然選択にさらされると予想されます。つまり、生殖適応度を変化させない変化は、世代から世代へと容易に受け継がれ、時間の経過とともに一般的になる可能性があります。逆に、適応度の低下をもたらす変化は、淘汰選択にさらされる可能性が高く、集団におけるその変異の頻度が時間とともに減少することにつながります。
適応度への影響は、遺伝的変異の頻度に影響を与えるいくつかの力のうちの1つに過ぎません。例えば、新しい変異は、定義上、集団に広がり、選択圧にさらされる時間があるまでは稀です。さらに、民族集団の歴史と移動パターンは、遺伝的変異の動態を時間とともに劇的に変化させる可能性があります。ただし、人種や民族性が社会的な構成物として、一般的な分散を伴う遺伝子研究を複雑にし、しばしば誤った発見や結論につながる可能性があることに注意することが重要です。
稀な変異は、発達の初期に致死的な障害や、生殖能力を低下させたり、生殖適応度を低下させたりする障害に寄与すると推定されます。逆に、一般的な遺伝的変異は、人生の後半に現れる障害や、適応度に負の影響を与えない障害に寄与すると推定されます。一般的な変異は、特定の遺伝的変異の正の効果(この場合はマラリア耐性)が負の結果を相殺する可能性がある状態(例:鎌状赤血球症の形質)にも関与すると推定されます。遺伝子変異の正と負の効果が競合する影響は、バランス選択と呼ばれます。
ヒト遺伝学の分野は、単一の稀な遺伝子変化が疾患や症候群を引き起こしたり、そのリスクを劇的に増加させたりする疾患において、計り知れない成果を上げてきました。しかし、複数の遺伝子変異が疾患の発現に寄与する障害を明確にするという課題は、依然として困難なままです。一般的な複合疾患の遺伝学を説明するために、2つの代替的な、しかし相互に排他的ではないパラダイムが出現しました。一般的な変異:一般的な疾患モデルと稀な変異:一般的な疾患の代替モデルです。
前者は精神疾患で主に支持されてきました。例えば、統合失調症、うつ病、双極性障害、自閉症は、それぞれが穏やかな効果を持ち、環境要因と相互作用して生物学的閾値を超える複数の一般的な遺伝的変異の複合効果から生じると広く考えられています。これらの疾患や他の複雑な疾患では、モデリングと初期の実験的証拠の両方が、特定の疾患に単一の表現型(比較的わずかな変動)があると仮定した場合、主要な効果を持つ単一遺伝子の寄与を本質的に排除しています。集団内の変動のほとんどは一般的な変異によって運ばれるため、一般的な疾患はこの基礎となる遺伝的アーキテクチャを反映する可能性が高いと考えられています。
代替モデルは、比較的大きな効果を持つ多くの個々に稀な変異が単独で、または組み合わされて、一般的な疾患のリスクに寄与すると提唱しています。この仮説は、早期発症の障害や生殖適応度を変化させる障害にとって直感的であるように思われます。バランス選択がない場合、たとえこれらが原因ではなく寄与的なものであっても、大きな効果を持つ対立遺伝子は稀である可能性が高いと予想されます。さらに、いわゆる散発的またはde novoの変異が重要な役割を果たした障害では、稀な突然変異の有意な負担が予想されます。実際、最近の研究では、自閉症スペクトラム障害(ASD)に罹患した子供におけるde novo突然変異(ほとんどはde novoミスセンス変異とde novo遺伝子破壊の可能性のある突然変異)が、診断された症例の10%以上を占める可能性があることが示されています。
これらの2つの可能性は相互に排他的ではありません。研究が進むにつれて、一般的な変異と稀な変異の両方が、多くの一般的な精神疾患の可変的な発現に寄与することが判明する可能性が高いです。しかし、この区別は現在の遺伝子研究にとって非常に重要です。なぜなら、遺伝子の原因的または寄与的な役割を示すために採用される方法論は、稀な遺伝的変異と一般的な遺伝的変異の寄与を検出する能力に関して大きく異なるからです。さらに、統合失調症やASDsのような複雑で極めて多様な表現型を持つ疾患は、稀な変異と一般的な変異の様々な組み合わせによって引き起こされる可能性があり、これが、少なくとも部分的には、前述の病態の広いスペクトルを説明することになるでしょう。
ゲノムの機能
ヒトゲノムの配列が解明された今、研究は30億個のヌクレオチドがどのように協調して機能するかに焦点を当てています。ヒトゲノムは、サイズ、遺伝子数、またはタンパク質をコードするゲノムの割合の点で、他の種と比較してほとんど特筆すべき点がないことを考えると、ヒト脳の能力は、比較的限られた数の遺伝子の多様性によって、または生のDNA配列以外の要因によって強力に影響を受けるに違いないと推定しなければなりません。ゲノムの脳特異的な機能的構成要素の特性評価と、これらの要素における遺伝的多様性の結果は、来るべき時代において精神医学遺伝学の主要な関心事となることは間違いありません。より広く言えば、このような理解は、ホモ・サピエンスを種として定義するユニークな分子変化を特定することになるでしょう。
最近の1970年代初頭まで、ヒト脳の発達を形成する上での遺伝子調節の重要性は疑問視されていました。ヒトと他の近縁種との違いの起源に関して、いくつかの仮説が検討されていました。種分化は、小規模な配列変化(DNAまたはタンパク質配列の変化)、調節変化(制御遺伝子または他の類似の要素からの変化)、または大規模な配列変化(遺伝子およびゲノム重複)の結果であると考えられていました。1975年、メアリー=クレア・キングとアラン・ウィルソンは、44個のヒトとチンパンジーの遺伝子を配列決定して比較した論文を発表しました。多くの人が驚いたことに、これらの配列の大部分はチンパンジーとヒトの間でほぼ同一であり、特定された小規模な変化のほぼすべてが同義でした。さらに、転写産物の遺伝子およびゲノム組織は、両種間で本質的に変化していませんでした。これらのデータは、コーディング配列もゲノム組織も、両種間の違いを説明する可能性が低いことを初めて示しました。
キングとウィルソンの研究は、遺伝子発現を調節する遺伝子周辺の配列が、皮質構造の劇的な違いを含む重要な進化的変化を説明するという代替的な結論を支持しました。この研究はまた、ヒトゲノムと近縁種のゲノムの配列決定だけでは、それらの生物学における主要な違いを理解するのに十分ではないことを示唆しました。これらの結論は大部分が裏付けられています。脳機能を理解するためには、遺伝子配列が存在するかどうかという問題は、その生物においていつ、どのように転写産物が活性であるかを決定することほど重要ではないことが明らかです。自閉症のような高度に分岐した神経発達障害の場合、脳の初期発達段階において、ASD関連遺伝子の独特な時空間発現パターンが存在することが示されています。これらの遺伝子の異常な時空間発現は、発達上の障害に容易に寄与し、自閉症を引き起こす可能性があります。
キングとウィルソンの研究のさらなる確認は、最近、旧世界のサル(Old World Apes)のゲノムを入手したことで得られました。すなわち、ヒトで急速な進化を遂げたいくつかのゲノム領域、つまりヒト加速領域(HARs)が特定されました。HAR1は新皮質の発生中に発現する遺伝子であり、HAR2(HACNS1とも呼ばれる)はヒト特異的な発達エンハンサーである一方で、他の721個のHARは配列の保存スコアを比較することで特定されました。驚くべきことに、それらの92%は非コーディング遺伝子であり、最近のヒトゲノム進化における調節配列の重要性を強調しています。
転写とトランスクリプトームの調節
おそらく、真核生物の複雑性への最大の貢献は、RNAスプライシングに由来します。これは、遺伝子のエキソンを複数の方法で組み合わせることができるプロセスです(図1.18-8)。
遺伝子がエキソンとイントロンの両方として転写されるという発見は、1977年にアデノウイルス遺伝子ヘキソンを研究することによって得られました。この発見がヒトの進化に与える影響はすぐに認識されました。つまり、DNAの単一の領域が様々な転写産物と複数のバージョンのタンパク質につながる可能性がありました。これらは、元の転写産物の有用性を犠牲にすることなく、新しい機能を生成する可能性のある進化的選択の標的となり得ました。したがって、トランスクリプトームの「宇宙」は突然はるかに大きくなりました。
RNAスプライシングの間、エキソンは成熟したメッセージに残されることも、様々な組み合わせで除去されることも可能であり、単一のpre-mRNAから多様なmRNAの配列を作り出します。このプロセスは代替RNAスプライシングと呼ばれます。1994年までに、代替スプライシングを示すヒト遺伝子の数は1%から5%と推定されていました。しかし、これらの推定値は、配列データの入手可能性が乏しいため不正確であることが知られていました。この問題の解決への関心は、HGPの一部が遺伝子構造と遺伝子発現の関係の研究に充てられることにつながり、**相補的DNA(cDNA)と発現配列タグ(EST)**の2つの方法が利用されました。
両方の方法は、mRNAが最初に処理されることに依存しており、これによりすべてのイントロンが除去され、初期の転写産物に長いポリAテールが追加されます。これらのポリAテールの存在は、相補的なポリT配列を詰めたカラムを通して抽出された総RNAを流すことによって、転写産物を結合および分離する手段を提供します。分離されると、一本鎖mRNAは、逆転写酵素と呼ばれる酵素を使用して相補配列を作成する反応のテンプレートとして機能します。これによりcDNAが作成され、その後配列決定することができます。ESTはcDNAの小さな断片であり、cDNAを消化し、細菌断片と配列を結合して配列を識別するためにクローニング技術を使用するため、わずかに異なります。作成および分析されると、これらのESTおよびcDNAライブラリは、ゲノム内の大量の代替スプライシングを確認し、遺伝子内では一部のエキソンが常に使用される(構成的にスプライシングされる)一方で、他のものは非常に稀で、体内の特定の時間または場所に特異的である(選択的にスプライシングされる)ことを示しました。
これらの配列ライブラリが作成されているのと同時に、研究者が遺伝子発現とスプライシングをより明確に確認できる他の技術も出現しました。1995年、パット・ブラウンらは、マイクロアレイを作成することで、植物シロイヌナズナの45個の遺伝子の発現を並行して初めて測定しました。彼らは、45個の遺伝子に相補的な配列をガラス製顕微鏡スライド上の小さなスポットに配置し、2つの異なるシロイヌナズナサンプルからmRNAを分離しました。45個の各遺伝子の相対発現は、スライド上の各スポットで決定された蛍光強度に基づいて、2つのサンプル間で決定することができました。

図1.18-8. 真核生物遺伝子における選択的スプライシング
この図は、細胞が環境条件や発達段階に応じて、特定の多エキソン遺伝子の異なるバリエーションを生成できることを示しています。これらの異なるスプライシングバリアントは、その後、遺伝子のタンパク質のわずかに改変されたバージョンを作り出します。
この時以来、一連の技術的および方法論的発展により、マイクロアレイ上に配置できるプローブ(スポット)の密度と特異性が驚くほど急速に増加しました。現在の技術では、数百万の配列を単一のスライド上に配列することが可能であり、生物内および生物間の遺伝子発現の包括的かつグローバルな研究の基礎を提供しています。数十万ものcDNAとESTが単離されており、現在の推定では、ヒト遺伝子の少なくとも80%が選択的スプライシングを受けるとされています。
生物のサイズや遺伝子数は一般的に生物の生物学的な洗練度とは相関しませんが、選択的スプライシングの場合はそうではありません。最も基本的なレベルでは、この区別は原核生物(選択的スプライシングがない)と真核生物(選択的スプライシングがある)との対比によって明確になります。また、高等生物においては、スプライシング率の増加がより大きな複雑性に対応するといういくつかの証拠もあります。例えば、酵母では、ほとんどの遺伝子(約96%)にイントロンがなく、スプライシングイベントはごくわずかしか発見されていません。さらに、最近の証拠は、無脊椎動物(ショウジョウバエでは50%から60%)は脊椎動物(ヒトでは80%)よりも選択的スプライシングを受ける遺伝子が少ないことを示しています。
ヒトゲノムにおける選択的スプライシングの総量は依然として疑問ですが、脳が人体内のどの組織よりも最も多くの量を占めることはすでに明らかです。この観察は、選択的スプライシングとその調節が、ヒト脳が他の近縁種とこれほど異なる理由を理解する上で、また精神疾患の神経生物学を理解する上で非常に重要である可能性を示唆しています。
実際、選択的スプライシングのプロセスは、遺伝子が加速された進化的変化を遂げる機会を与えます。ほとんどの遺伝子は強い負の選択圧にさらされており、これは特に生物の初期発達にとって重要である場合、それらが変化しにくいことを意味します。遺伝子の稀な産物の形成につながる選択的スプライシングは、この負の選択圧を緩和する進化的近道となり得ます。主要な(またはより一般的な)形態が十分に存在するため、新しい、マイナーなスプライスフォームは同じ程度の負の選択を受けず、生物にとって新しい機能を獲得するための「中立空間」さえ作り出す可能性があります。
このプロセスの十分な証拠はDNA配列内に見つけることができます。マウスとヒトゲノムの間で、特定の遺伝子の主要な形態とマイナーな形態のエキソン構成は、大部分が保存されています。主要な利用エキソンは通常、マイナーな利用エキソンよりも高い配列保存度を示し、前者に対するより強い負の選択を示唆しています。種間比較は、マイナーな利用エキソンが進化的に最近の追加であることを示しており、遺伝子の基本的な生物学的機能が新しいスプライスバリアントの追加によって増強され得るという仮説を裏付けています。マイナーな利用エキソンが新しい重要な機能を果たし始めると、その保存レベルがより古いエキソンのそれと急速に近づくという強い証拠があります。これらの現象は、新しいスプライスバリアントが時間的または空間的に高度に限定されている場合でも起こり、これはヒト脳で十分に実証されています。
研究者が、遺伝子の最も高度に保存された領域、またはタンパク質配列の最も高度に保存されたアミノ酸に影響を与える変化に焦点を当ててきたことはよくありました。これは、これらが転写産物の最も重要な機能の基礎となるという考えに基づいています。最近の種間比較とスプライス多様性に関する知識の深化は、あまり保存されていない配列や、空間的または時間的に限定されたマイナーなスプライスバリアントのみを変化させる変化も非常に重要である可能性があることを示唆しています。
スプライス多様性の重要性の理解は、その調節の重要な性質を指し示しています。ゲノムの他の多くの側面と同様に、プロセスのメカニズムは不完全に理解されています。イントロンの境界を定義する典型的なスプライス部位が存在することは長らく認識されてきました。5’スプライス部位は通常GT配列を持ち、3’部位はほとんどの場合AG配列を持ちます。しかし、これらは既知のイントロンの約2%には存在しないため、絶対的なものではありません。遺伝子スプライシングの調節は、遺伝子の近くまたは内部の配列によっても影響されることが認識されてきました。これらには、エキソン性スプライシングエンハンサー(ESEs)とエキソン性スプライシングサイレンサー(ESSs)が含まれます。これらは、スプライシングの増殖または減衰を調節するエキソン内に含まれる特定の配列です。これらの配列の変化は、タンパク質のアミノ酸組成を変化させることなく、遺伝子機能に深刻な影響を与える可能性があります。これらのモチーフに関与する同義配列変異、すなわちヌクレオチド配列は変化させるがアミノ酸配列は変化させない変異が、場合によっては深刻な発達障害につながることが示されています。
遺伝子スプライシングに関する知識の蓄積に加えて、トランスクリプトームを調節する他のいくつかの側面もよく特徴づけられています。個々の遺伝子は転写因子によって調節され、これらは遺伝子の活動のタイミングと程度を決定します。転写因子は、遺伝子の上流にある特定の配列、TFBSs(転写因子結合部位)またはREs(応答要素)に結合します(図1.18-9)。ほとんどの転写因子は、標的特異性を付与するDNA結合ドメインと、その調節機能を付与する活性化ドメインで構成されています。
RNA編集
RNA編集は、トランスクリプトームを制御し多様化させるもう一つの方法です。RNA編集は、ゲノムのDNAを変化させることなく、生物の情報内容を改変できるため、最も珍しい転写後RNA修飾の一つです。これは、ADAR型酵素によって媒介され、アデノシン(A)を脱アミノ化してイノシン(I)に変化させます。このイノシンは、スプライソソームとリボソームの両方によってグアノシン(G)として認識されます(図1.18-10)。
この種のトランスクリプトームの多様化は、遺伝子重複/突然変異や選択的スプライシングの代わりに、効率性と低エネルギー消費のためか、多くのケースで進化的な変化に貢献してきました。RNA編集は、コード配列、miRNA結合部位に変化をもたらし、新規スプライシング部位を作り出す可能性があります。
最近まで、編集されたヒト遺伝子はごく少数しか発見されておらず、そのほとんどが中枢神経系で重要な役割を担っていました。脳は、AMPA、HTR2C受容体、ナトリウムチャネル、カルシウムチャネルなど、RNA編集イベントの最も多才なレパートリーを示します。ヒトは、げっ歯類、アカゲザル、チンパンジーと比較して、AからIへの編集量が最も多く、これがヒトの特性に貢献している可能性があります。
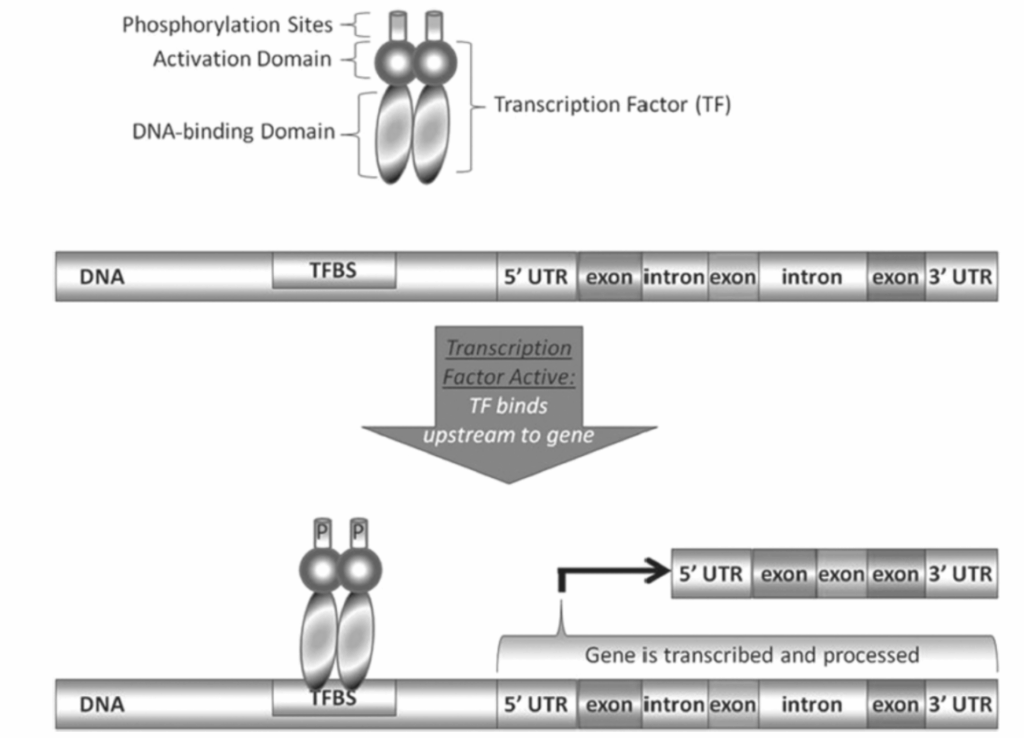
図1.18-9. 転写因子による遺伝子制御。転写因子は、その活性化ドメインが通常リン酸化(TF上のPサイト)によって活性化されると、DNA結合ドメインが活性化され、適切な遺伝子の前にある**結合部位(TFBS)**を見つけます。結合すると、その遺伝子が活性化されます。
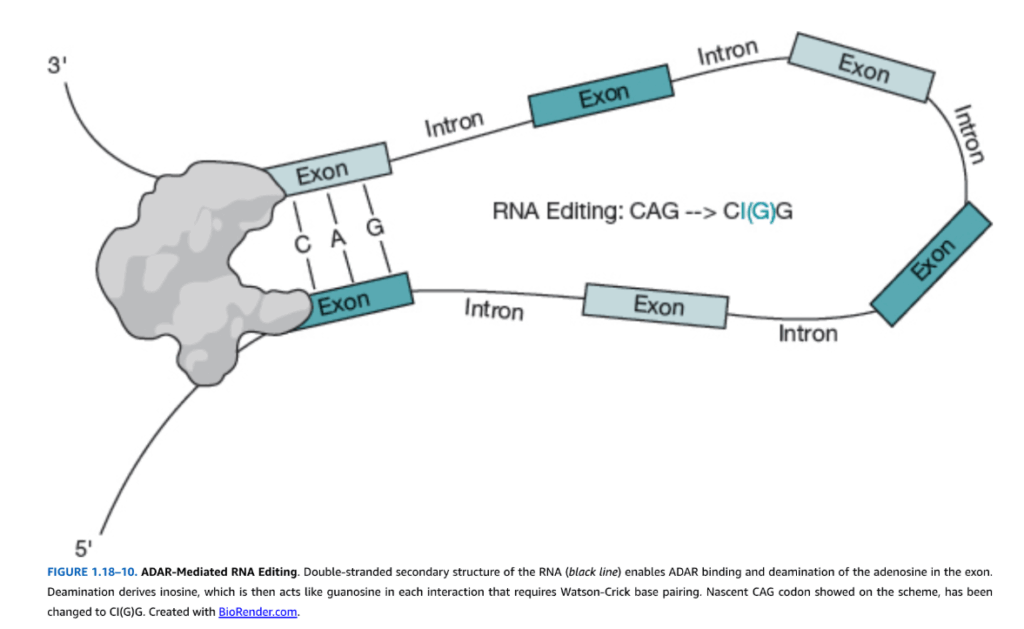
図1.18-10. ADARによるRNA編集。RNAの二重らせん二次構造(黒線)は、ADARの結合とエクソン中のアデノシンの脱アミノ化を可能にします。脱アミノ化によりイノシンが生成され、これはワトソン・クリック型塩基対形成を必要とするあらゆる相互作用においてグアノシンのように振る舞います。図に示されている新生CAGコドンは、CI(G)Gに変化しています。BioRender.comで作成。
RNA編集は、二本鎖二次構造(dsRNA)を形成するRNA上でほぼ排他的に発生します。Aluエレメントは、その特定の配列のために、RNA編集の完璧な標的となります。脳機能に関与する非常に多くの遺伝子(カルシウム、ナトリウム、カリウムチャネル;GABA、セロトニン、グルタミン酸受容体)が編集を受けるという事実から、RNA編集は脳のトランスクリプトームの「微調整役」であると考えられています。
最もよく知られている真核生物の調節要素の1つはTATAボックスです。これは、TATA配列が遺伝子の上流に存在することからそう名付けられました。このモチーフは、転写開始を可能にするタンパク質複合体を結合します。個々の遺伝子のコアプロモーターは、すべての要素を含んでいる必要はありません。多くのプロモーターはTATAボックスを欠いており、その代わりに機能的に類似のイニシエーター要素(INR)を使用します。これらの配列は、ゲノム中のタンパク質コード遺伝子の数に関連しています。生物のコード遺伝子の数を決定することは、単にコード領域の近くにあるプロモーター中のTATAボックスとINRの数を数えることと同じです。
TATAボックスのように遺伝子のすぐ上流にある配列は、その遺伝子の近くにあり、同じDNA鎖上にあることから、シス調節因子(「同じ側にある」という意味)と呼ばれます。DNAの別の鎖から、またはゲノムの遠隔領域から遺伝子に作用する調節要素、TFBS、または遺伝子産物は、トランス調節要素と呼ばれます。
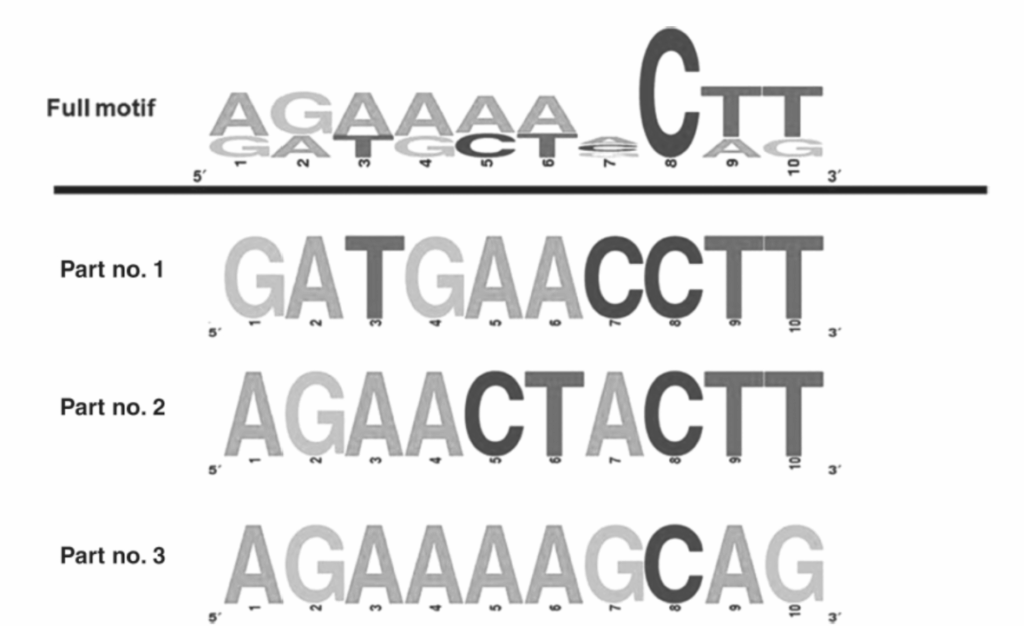
図1.18-11. 転写因子結合部位(TFBS)のコンセンサス配列の構築。多くの遺伝子の上流にあるTFBSの場合、その特定のTFの結合を可能にする複数の異なる機能モチーフ(パート#1、#2、および#3)が存在することがよくあります。これら3つの10bpの配列を統合すると、位置8(常にシトシン(C))を除くすべてのヌクレオチドで縮重したコンセンサスモチーフが作成されます。
ヒトゲノムが解読された後、この単純な調節の二分法(シスとトランス)はより複雑になりました。最もよく理解されているプロモーター要素(TATAボックスとINR)は、ヒト遺伝子の54%のプロモーター領域にのみ存在し、プロモーターは転写開始部位(TSS)のどちらかの方向に100 bp以内に定義されます。これは、これらの遺伝子の発現と機能を調節するために未知のプロモーター配列が存在するか、または遺伝子調節の多くがトランス調節要素で起こることを示唆しています。現在、多くのバイオインフォマティクス研究がこの問題に焦点を当てています。
現在、10を超えるコアプロモーター要素が予測されていますが、これらの要素がすべての、あるいはほとんどの遺伝子に普遍的に存在するという期待はもはやありません。むしろ、遺伝子は大規模な同時グループで活性化されるか、複数の活性化の波で現れ、これらの共発現遺伝子は、特定の時間または場所に発現を制限する特定の調節要素とモジュールを持つことができます。大規模な共発現遺伝子のセットが見つかると、これらの遺伝子の上流領域を調べて調節要素の配列を特定することができます。
共発現遺伝子のセットの場合、これらのすべての転写産物の上流で全く同じ調節配列を特定することは一般的ではありません。TFのDNA結合ドメインの柔軟性のため、遺伝子調節において許容されるある程度の配列多様性がしばしば存在します。多くのTFBSの研究により、結合部位の特定のヌクレオチドが重要である一方、他のヌクレオチドは変動する可能性があることが示されています。これらの部位を多くの遺伝子間でアラインメントすると、TFBSのコンセンサス配列またはコンセンサスモチーフが特定されます(図1.18-11)。
現在、数百の転写因子がChIP(クロマチン免疫沈降法)と呼ばれる技術で研究されています。TFの調節要素への動的な結合プロセスは、クロスリンクとして知られる化学プロセスを介して、結合したTFをそれらの同族の調節部位に固定することによって、一瞬にして凍結されます。ゲノムDNAの一部はTFと結合しており、このクロスリンクを保持するように断片化されます。既知のTFに特異的な抗体を使用することで、結合したゲノム配列を特定し、ゲノムの残りの部分(未結合の断片)から分離し、特性を評価することができます。これらの結合した断片は、特定のTFがどこで機能しているかを正確に特定するために直接シーケンスすることも、異なる色素で標識してマイクロアレイで研究することもできます。後者の方法はChIP-chip(チップ上のクロマチン免疫沈降)として知られています。
表1.18-6.
最も主要なヒストンマーカーに対するリジンのモノ、ジ、トリメチル化、およびアセチル化が遺伝子発現に与える影響
はい、ヒストンマーカーとその修飾が遺伝子発現に与える影響を表形式でまとめます。
承知いたしました。画像として提供していただき、ありがとうございます。これで、各ヒストンマーカーと修飾の関係性が明確に理解できました。
画像の内容を元に、正確な表を日本語で作成します。
ヒストン修飾の種類と遺伝子発現への影響
| ヒストンマーカー | モノメチル化 | ジメチル化 | トリメチル化 | アセチル化 |
| H3K4 | Activation | Activation | ||
| H3K9 | Activation | Repression | Repression | Activation |
| H3K14 | Activation | |||
| H3K27 | Activation | Repression | Repression | Activation |
| H3K79 | Activation | Activation | Activation | |
| H3K36 | Activation | |||
| H4K20 | Activation | |||
| H2BK5 | Activation | Repression |
これらの実験により、ヒトゲノムには非常に多くの調節の複雑性が存在することが明らかになりました。遺伝子の転写が開始されるTSSの位置は、最初のエクソンから数十万塩基(>100 kb)離れていることもあります。また、多くの遺伝子のこれらの調節要素、TSS、およびTFBSは、それぞれの遺伝子の隣ではなく、イントロン内、あるいは全く異なる遺伝子内に見られます。
これらの調節要素は、遺伝子が個別の要素として機能することを可能にすることもありますが、時には、これらの調節モチーフによって遺伝子のグループが結合することもあります。トランススプライシングの観察例もあり、ある遺伝子のエクソンが全く異なる遺伝子のエクソンと結合し、2つの独立したタンパク質の一部または全部の要素を含むキメラmRNAとキメラキメラタンパク質が生成されます。
遺伝子調節を理解する上で重要な点は次のとおりです。ゲノム内のいかなる調節要素も、隣接する遺伝子を調節していると仮定すべきではありません。複数の遺伝子が単一の遠隔の調節要素によって同時に活性化されることがあります。そして、単一の遺伝子は常に単一の遺伝子であるとは限らず、比較的遠い距離であっても、別の遺伝子の一部または全部と(必要に応じて)結合することができます。
エピジェネティクス
遺伝子調節のもう一つの領域は、ゲノム配列の外側にあるエピゲノムに存在します。エピジェネティックな調節とは、ヌクレオチド配列を変化させない、ゲノムにおけるあらゆる遺伝性の変化のことです。これらの変化は、メチル化として知られるDNAへの化学修飾、またはヒストンのタンパク質複合体への化学変化によって起こります。ヒストン修飾ははるかに多く、リン酸化、アセチル化、メチル化、ユビキチン化がDNA-タンパク質複合体の立体構造を変化させることで、遺伝子または遺伝子群の活性を調節します。これは、転写因子へのDNAの利用可能性、および遺伝子発現を阻害または促進する他の分子の動員に影響を与えます。
DNA上のエピジェネティックマークは、特定のメチルトランスフェラーゼによるシトシンのメチル化からなり、それによって5-メチルシトシン(5mC)が生成されます。5mCの存在は、隣接するCGタンデム配列であるCpGサイトを含むゲノム領域で優勢です。遺伝子プロモーターに位置するCpGサイトのシトシンメチル化は、転写的にサイレンシングされた遺伝子を特徴づけるものです。DNAメチル化は可逆的なプロセスであり、5mCの酸化生成物である5-ヒドロキシメチルシトシン(5hmC)は、5mCがシトシンに戻る際のN中間体であると考えられています。生殖細胞における5hmCの存在は、配偶子の生産に脱メチル化(エピジェネティックコードのリセット)が必要であるため、このモデルを支持します。さらに、5hmCは中枢神経系で高濃度を示し、5hmCが脳におけるスプライシングと遺伝子発現の調節に役割を果たしていることを示唆する新しいデータがあります。
重要なヒストン修飾とその遺伝子発現への影響は、表1.18-6に示されています。
おそらく、遺伝子抑制の最も信頼できる兆候は、遺伝子プロモーターにおけるH3K27Me3の存在、および特定の位置でのヘテロクロマチンの存在を示す強力なマークであるH3K9Me2/3です。対照的に、プロモーターにおけるH3K4Me3の存在は、開放的で活発に転写されているクロマチン領域の兆候であり、遺伝子全体の長さにおけるH3K36Me3マークも活発に転写されている領域の指標です。
エピジェネティックなプログラミングは、全身にわたる遺伝子機能の組織特異的調節と構造的および機能的特殊化を可能にします。エピジェネティクスは、同じDNA配列を共有する一卵性双生児間の違いを説明するのにも明らかに役立ちます。この非配列ベースの遺伝性変化は、受精時の親特異的な影響、つまりエピジェネティックインプリンティングにつながることもあります。これらのエピジェネティックマーカーは、親の生殖細胞(精子または卵子)で確立または「インプリント」され、生物の体細胞における有糸分裂を通じて維持されます。各個人は、卵子または精子を生成する際に、自分の親から与えられたエピジェネティックなシグネチャを再プログラムする必要があるため、自分の性別のエピジェネティックな特徴をインプリントします。プラダー・ウィリー症候群とアンジェルマン症候群は、異常なエピジェネティックインプリンティングに起因する疾患の例です。
エピジェネティックなプログラミングは、単一の遺伝子を調節するだけでなく、染色体全体の挙動を変化させることもできます。X染色体不活性化は、すべてのヒト女性に起こる正常なプロセスであり、すべての細胞で1つのX染色体が遺伝的にサイレンシングされます。これにより、用量補償が可能になり、X染色体にマッピングされたあらゆる遺伝子の発現量は、男性と女性で本質的に同じになります(男性は1本のX染色体しか持っていないにもかかわらず)。特定の細胞内でどの染色体が活性(X2)で不活性(X)であるかの選択は、通常はランダムなプロセスです。しかし、場合によっては、特定の染色体が不活性化される可能性が高く、レット症候群やその他のX連鎖性疾患の一因となります。
親由来効果とは、常染色体が母親または父親から伝達されたかどうかに基づいて疾患のリスクが伝達されることとして定義され、いくつかの一般的な精神疾患において役割を果たすと仮説が立てられています。この可能性が最も強く示唆される証拠は、母親から遺伝した染色体15qの重複が、父親から遺伝したものではないが、自閉症の症例のごく一部ではあるが有意な割合を占めるという観察です。
エピトランスクリプトーム
もう一つの複雑性の層は、RNAの塩基の修飾、すなわちエピトランスクリプトームから生じます。現在のデータベースには、112種類のRNA修飾がリストされています。そのうちの1つ、メチル-6-アデノシン(m6A)は、すべての生物の王国で発見されています。m6Aは、マウスとヒトの両方で3′ UTRと長いエクソンに豊富に存在します。m6Aが豊富な配列は進化的に保存されています。予備分析では、m6AがRNA編集を妨げる可能性があることが示されました。また、m6Aは脱メチル化されてアデノシンに戻るため、RNA編集を調節するメカニズムを構成する可能性があります。m6Aは概日リズムの調節に関与しており、おそらく概日周期調節遺伝子の核膜を通じた輸送に影響を与えることによって関与している可能性があります。さらに、m6Aが豊富な転写産物の翻訳動態の調節に役割を果たす可能性があります。これらの機能的関係は、RNAの転写後修飾が転写産物の特性と機能を変化させ、いわゆるエピトランスクリプトームを生成することを示唆しています。
タンパク質調節とプロテオーム
遺伝子発現が複数の段階で調節されるのと同様に、コード遺伝子によって指定されるタンパク質産物もまた調節されます(表1.18-7)。20種類以上の既知のタンパク質修飾が存在します。これらの修飾には、特定のアミノ酸の化学的修飾、別のアミノ酸への交換、他の生化学的機能基の付加、より大きな分子(例:脂質や炭水化物)の付加、またはタンパク質への構造変化が含まれます。タンパク質が完全に形成され、生化学的に活性になった後でも、選択的に分解の標的となることがあります。
これらの種類のタンパク質修飾は、ほとんどの神経伝達物質がシナプス膜やシナプス間隙を、このタスクを達成するために特別に作られたタンパク質によって輸送されるため、神経伝達において重要な役割を果たします。例えば、シナプス膜を介してドーパミンを輸送するタンパク質であるDATは、各細胞における必要な機能に特異的な一連の翻訳後変化を受けます。
表1.18-7. ゲノム、トランスクリプトーム、およびプロテオームにおける調節メカニズム
| 調節メカニズム | 段階 |
| 対立遺伝子排除(遺伝子の一方の対立遺伝子をインプリンティングし、他方のみを発現させる) | エピジェネティック |
| X染色体不活性化 | エピジェネティック |
| クロマチンリモデリングと化学的改変(メチル、アセチル、リン酸) | エピジェネティック |
| 短距離の細胞間シグナル伝達 | 転写前 |
| 組織特異的転写因子の結合 | 転写前 |
| 競合的阻害因子の結合 | 転写前 |
| 誘導性遺伝子における応答要素へのホルモン結合 | 転写前 |
| 異なる転写開始部位、停止部位、プロモーターの使用 | 転写後 |
| 同一遺伝子内の選択的スプライシング | 転写後 |
| 組織特異的RNA編集 | 転写後 |
| 翻訳制御メカニズム | 翻訳後 |
| siRNAによる干渉と分解 | 翻訳後 |
| 異なる遺伝子間のトランススプライシング | 翻訳後 |
| タンパク質分解的切断 | 翻訳後 |
| タンパク質のより大きなタンパク質複合体への統合 | 翻訳後 |
| 糖鎖付加 | 翻訳後 |
| 分解の標的化(ユビキチン化) | 翻訳後 |
| 化学的改変(メチル、アシル、リン酸、または硫酸基) | 翻訳後 |
| 重金属の付加 | 翻訳後 |
これらの変化を各タンパク質について測定し理解するために、プロテオミクスという新しい分野が生まれました。この分野では、個々のタンパク質の様々な化学修飾、およびこれらのタンパク質が互いにどのように相互作用してより大きな細胞複合体や機構を形成するかを調べます。プロテオームの構成要素は集中的な研究の対象となっており、例えば、シナプス内のすべての活性な相互作用タンパク質はシナプスプロテオームと呼ばれ、脳内のタンパク質は脳プロテオームと呼ばれます。これらのすべてのタンパク質特性評価と相互作用には、広範な実験的サポート、大規模なデータセット、および重複する実験方法が必要です。
質量分析法は、サンプル中に存在するすべてのタンパク質を迅速にカタログ化するのに役立ちます。例えば、シナプスプロテオームには約1,000のタンパク質が含まれています。質量分析法と組み合わせた高速液体クロマトグラフィー(HPLC)は、脳の全プロテオームを解明する能力を持っています。二次元(2D)電気泳動は、タンパク質相互作用を研究するために使用されます。このアプローチでは、ゲル中でタンパク質を互いの経路に沿って移動させ、移動パターンの変化が直接的な相互作用を示唆します。酵母ツーハイブリッド実験は、タンパク質結合パートナーを特徴付けるための別のアプローチです。この場合、2つの対象タンパク質が酵母細胞にクローン化されます。各タンパク質はハイブリッドであり、一方は最初のタンパク質と特定の転写因子のDNA結合ドメインを含み、もう一方は2番目のタンパク質と同じ転写因子の活性化ドメインを含みます。2つのタンパク質が相互作用すると、転写因子は標的レポーター遺伝子を活性化するために必要な両方のコンポーネントを持つことになり、通常、このレポーター遺伝子はアッセイしやすいように設計されています。より最近では、発現マイクロアレイに似たプロテインチップが作成されています。これらのチップは、数千のタンパク質を顕微鏡スライド上に配置し、単一のサンプルで洗い流すことで、タンパク質と照会されたプロテオームの残りの部分との間のすべての相互作用を検出します。
これらのすべての方法は、ヒトゲノムの約25,000の遺伝子によって作られるタンパク質の発見と特性評価を可能にしました。現在の推定では、この比較的少ない数のコード遺伝子から、最大200万種類の異なるタンパク質が生成される可能性があります。これらのすべてのタンパク質を正確にカタログ化するという課題に取り組むために、Human Proteome Initiative(HPI)やHuman Proteome Organization(HUPO)など、いくつかのグループが発足しています。
要約すると、ゲノムを調節する複雑な機構は、転写前調節(転写因子の機能によって例示される)、転写後調節(miRNAおよびsiRNAによって例示される)、翻訳後調節(タンパク質の化学的修飾およびプロセシングによって例示される)、およびエピジェネティック調節の4つのレベルを包含すると考えることができます。
これらのプロセスがヒト脳の複雑な機能にどのように貢献し、精神疾患にどのように寄与しうるかを理解するという概念が十分に困難でないとしても、トランスクリプトームとプロテオームに内在する多様性の天文学的な可能性を考慮するだけで十分です。ヒトゲノムはわずか30億文字の長さですが、複数のエクソンを持つすべての遺伝子において、可能な転写産物の数は2n-1の割合で増加します。したがって、2つのエクソンを持つ遺伝子は、2つのエクソンそれぞれ、または両方を組み合わせた生成物にスプライスされ、合計3つの可能な転写産物となります。同様に、3つのエクソンを持つ遺伝子は、3つのエクソンの一つ、または他の4つの組み合わせ(1-2、2-3、1-3、1-2-3)にスプライスされ、合計7つとなり、以下同様です。この式をヒトゲノムで最も多くのエクソンを持つ遺伝子(チチン、234エクソン)に適用すると、この1つのコード単位が理論的に2.76 × 10^70種類の異なる転写産物に結合できることがわかります。さらに、ヒトプロテオームには約200万のタンパク質が存在することを考えると、4兆通りのタンパク質間相互作用が存在します。合計すると、これは約7.0 × 10^82通りの可能な遺伝子産物とその後の相互作用を検討することになります。比較として、宇宙の原子の数は約4.0 × 10^80と推定されています。これらの数字は理論的な近似値ですが、ヒトゲノムの広大な潜在的複雑性と、それを理解するための洗練された手法の必要性を示しています。
神経精神疾患におけるファーマコゲノミクス
ファーマコゲノミクスは、個々の患者の治療を最適化するという長期的な目標を掲げ、薬剤への反応に影響を与える遺伝子変異を特徴づける取り組みです。ファーマコゲノミクスの検査は、腫瘍学、感染症、麻酔学、消化器科、心臓病学、婦人科、リウマチ学、神経学、精神医学など、様々な臨床分野で既に確立されています。米国食品医薬品局(FDA)は、350以上の薬物と遺伝子の組み合わせをカバーする75以上の遺伝子を対象とした、薬剤表示におけるファーマコゲノミクスバイオマーカーの表を管理しています。
精神医学において、ファーマコゲノミクスは、向精神薬の代謝速度に影響を与えるP450ミクロソーム酵素をコードする遺伝子変異を特徴づけることで、最も大きな影響を与えてきました。これらの検査は、薬物血中濃度測定の代替として臨床的に適用されており、臨床反応の欠如が急速な薬物クリアランスに起因するかどうかを特定し、より高用量の薬物治療が必要かどうかを判断するために利用されています。これにより、薬物の副作用に敏感な遅代謝者も特定できる可能性があります。現在までのところ、向精神薬の薬力学的作用に関するファーマコゲノミクス研究は、日常的な臨床使用のための十分な妥当性や説明力を持っていませんが、将来的にはより一般的になる可能性があります。
システムとしての脳
システム生物学の分野は、生物学的システムの創発的特性に焦点を当てています。科学者がゲノム、トランスクリプトーム、プロテオーム、および行動などの脳の他の創発的特性の相互作用を評価できる洗練された計算アプローチの開発への関心が高まっています。
システム生物学は、ヒトゲノム、トランスクリプトーム、プロテオームの完全な機能的注釈を目的とした大規模な国際共同研究によって生成される、非常に大規模なデータセットに対して特に強力なアプローチです。これらの取り組みは、本章で議論されているすべての種類のデータ(調節配列、それに結合する転写因子、発現遺伝子(コードおよび非コード)、他の酵素によるその後の修飾、最終的なタンパク質産物、およびそのタンパク質と他のタンパク質との相互作用(タンパク質間相互作用[PPI]))の蓄積を可能にした技術進歩によって可能になりました。これらのデータは3つの情報源から得られます。(1) 転写活性、スプライシングの可能性、またはタンパク質相互作用の計算による予測。(2) 公開された科学レポートとデータベースから抽出された実験に基づく観察。(3) 世界中の公開された科学文献をスキャンし、関心のある特定の遺伝子やタンパク質との相互作用を報告するアルゴリズム(テキストマイニング)の使用。さらに、UniProtなどのタンパク質配列の大規模データベースがコンパイルされ、キュレーションされています。
過去数年間で、バイオインフォマティクスと計算生物学における大きな進歩により、生化学的または神経学的経路全体をモデル化し、経路の1つのセグメントの変化(変異したエクソンからシナプス可塑性の低下まで)を、他の考えられる相互作用する生物学的実体のすべてに追跡できるようになりました。この手法(パスウェイ解析として知られています)は、同じ経路内の他の遺伝子やタンパク質への予想される変化を予測します。Ingenuity、DAVID、KEGG、そしてCold Spring Harbor Laboratoryがヒト体内に存在するすべての相互作用(遺伝子-遺伝子、遺伝子-タンパク質、タンパク質-タンパク質)の機能モデルを構築する取り組みであるInteractomeなど、いくつかのツールが既にオンラインで一般公開されています。
表1.18-8. オンライン科学リソース
| 説明 | アドレス |
| National Center for Biotechnology Information | https://www.ncbi.nlm.nih.gov/ |
| Wellcome Trust Genome Research Center | https://www.sanger.ac.uk/ |
| The HapMap Consortium | https://www.genome.gov/10001688/international-hapmap-project |
| National Human Genome Research Institute | https://www.genome.gov/ |
| Protein Data Bank | https://www.rcsb.org/ |
| The ENCODE Project | https://www.encodeproject.org/ |
| UCSC Genome Bioinformatics Center | https://genome.ucsc.edu/ |
| Personal Genome Project | https://www.personalgenomes.org/ |
| Allen Brain Atlas from Mouse Brains | https://portal.brain-map.org/ |
| Functional Annotation of Genes | http://geneontology.org/ |
| The Human Protein Atlas | https://www.proteinatlas.org/ |
| Database and Research Tools for Proteins | https://proteinlounge.com/index.php |
| Modeling of Biomolecular Interactions | http://interactome.org/ |
最近、マウス脳の領域特異的発現解析をカタログ化した、アレン脳アトラスとして知られる並外れた公共リソースが作成されました。このデータベースには、現在、ほぼすべての遺伝子の発現パターンの三次元レンダリングが含まれています。マウスモデルは、ヒトの領域特異的脳発現を理解するための良い代理となり、この包括的なリソースの結果として、基本的に任意の関心のある遺伝子を他のすべての遺伝子と分析することができ、例えば、遺伝子グループ間の空間的および時間的相関やゲノム全体にわたる相関を調査することができます。これらのリソースやその他のリソースは、ワールドワイドウェブ上で一般公開されています(表1.18-8)。
将来の見通し
脳は人体で最も複雑な臓器であり、おそらく人類に知られている中で最も複雑な構造です。科学者はこれまで、脳を研究するためにこれほど多くのツールと技術を持っていたことはありませんでした。これらのアプローチは、生物学的システムに関する情報を収集し統合する比類のない能力を生み出していますが、臨床医や研究者が患者データを統合し、抽象的な予測を行うための正確な方法は、現在も開発途上です。
シーケンシング、マイクロアレイ、クローニングは不可欠な技術でしたが、すでに新しい、より高速で、よりハイスループットな技術によって凌駕されつつあります。現在、個々の患者のゲノムを分析し理解するために使用されているすべての方法は、特定の領域に制限されていますが、これは現在の技術の限界にすぎません。各患者のゲノムが安価かつ迅速にシーケンスできるようになれば、ポストゲノム時代のもう一つの革命が始まるでしょう。変異や構造異常を疑われる遺伝子や場所のみで探すのではなく、ゲノムのすべての場所で大小すべての遺伝的変異が検出されるようになるでしょう。
明らかに、各患者のDNAの完全な情報が得られることで、精神障害に寄与し、治療反応に影響を与え、環境ストレス因子の結果を媒介する遺伝的変異の割合を特定する努力が大幅に強化されるでしょう。最近の発見が、パーソナライズされたゲノム医療の開発に向けた最初の一歩をすでに告げていることは間違いありません。1,000ドル以下のゲノムは、この可能性を実現するための大きな一歩です。分子遺伝学からの莫大な恩恵は、DNA配列と構造変化の理解を大きく深めるでしょう。これらの重要な最初の一歩は、精神疾患の分子神経生物学を解明し、新しい治療法を開発する上で不可欠となるでしょう。
参考文献
An JJ, Gharami K, Liao GY, et al. Distinct role of long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell. 2008;134(1):175-187.
Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817-846.
Capra JA, Erwin GD, McKinsey G, Rubenstein JL, Pollard KS. Many human accelerated regions are developmental enhancers. Philos Trans R Soc Lond B Biol Sci. 2013;368(1632):20130025.
da Silveira WA, Fazelinia H, Rosenthal SB, et al. Comprehensive multi-omics analysis reveals mitochondrial stress as a central biological hub for spaceflight impact. Cell. 2020;183(5):1185-
1201.e20.
Drozda K, Müller DJ, Bishop JR. Pharmacogenomic testing for neuropsychiatric drugs: current status of drug labeling, guidelines for using genetic information, and test options. Pharmacother-
apy. 2014;34(2):166-184.
Dulai KS, von Dornum M, Mollon JD, Hunt DM. The evolution of trichromatic color vision by opsin gene duplication in New World and Old World primates. Genome Res. 1999;9(7):629-638.
ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799-716.
Frankish A, Harrow J. GENCODE Pseudogenes. Pseudogenes. Springer; 2014:129-155.
Gerstein MB, Bruce C, Rozowsky JS, et al. What is a gene, post-ENCODE? History and updated definition. Genome Res. 2007;17(6):669-681.
ens M, Nikolaus R. Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat Rev Genet. 2015;16(2):113-126.
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478):eaau6977.
Kapranov P, Willingham AT, Gingeras TR. Genome-wide transcription and the implications for genomic organization. Nat Rev Genet. 2007;8(6):413-423.
Kellis M, Wold B, Snyder MP, et al. Defining functional DNA elements in the human genome. Proc Natl Acad Sci USA. 2014;111(17):6131-6138.
Khaja R, Zhang J, MacDonald JR, et al. Genome assembly comparison identifies structural variants in the human genome. Nat Genet. 2006;38(12):1413-1418.
King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107-116.
Lander ES, Linton LM, Birren B, et al .; International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860-921.
Levy S, Sutton G, Ng PC, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5(10):e254.
Liu GE, Alkan C, Jiang L, Zhao S, Eichler EE. Comparative analysis of Alu repeats in primate genomes. Genome Res. 2009;19:876-885.
Mendel JG. Versuche über pflanzenhybriden. In: Verhandlungen des naturforschenden Vereines in Brünn 4. Abhandlungen; 1866:3-47.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA Methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635-
1646
Oldham MC, Horvath S, Geschwind DH. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc Natl Acad Sci U S A. 2006;103(47):17973-17978.
Paz-Yaacov N, Levanon EY, Nevo E, et al. Adenosine-to-inosine RNA editing shapes transcriptome diversity in primates. Proc Natl Acad Sci USA. 2010;107(27):12174-12179.
Pipes L, Li S, Bozinoski M, et al. The non-human primate reference transcriptome resource (NHPRTR) for comparative functional genomics. Nucleic Acids Res. 2013;41(D1):D906-D914.
Plotkin JB, Grzegorz K. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genetics. 2011;12(1):32-42.
Pollard KS, Salama SR, King B, et al. Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 2006;2(10):e168.
Pollard KS, Salama SR, Lambert N, et al. An RNA gene expressed during cortical development evolved rapidly in humans. Nature. 2006;443(7108):167-172.
Stolc V, Gauhar Z, Mason C, et al. A gene expression map for the euchromatic genome of Drosophila melanogaster. Science. 2004;306(5696):655-660.
Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291(5507):1304-1351.
Wang HY, Chien HC, Osada N, et al. Rate of evolution in brain-expressed genes in humans and other primates. PLoS Biol. 2007;5(2):e13
Wilson FH, Hariri A, Farhi A, et al. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science. 2004;306(5699):1190-1194
Zhang ZD, Paccanaro A, Fu Y, et al. Statistical analysis of the genomic distribution and correlation of regulatory elements in the ENCODE regions. Genome Res. 2007;17(6):787-797.
