
1.19 精神疾患の遺伝子マッピング研究
スコット・C・フィアーズ医学博士
遺伝学の分野は、20世紀初頭にグレゴール・メンデルの基本概念が再発見されたことから本格的に始まり、生物科学だけでなく、医学全体の不可欠な基礎として成熟してきました。世紀半ばのDNAの基本的な構造と特性の発見は、ヒトゲノムやその他無数の種の完全な配列の解読を含む、生命科学のあらゆる側面に関する現在の理解の指数関数的な加速をもたらしました。このような配列の大規模なデータベースは、21世紀の生物学者にこのすべての情報の機能的意義を解読するという課題を与えています。特に、配列のバリエーションが種間および種内の個体間の表現型のバリエーションにどのように寄与するかを決定することに注目が集まっています。ヒトにおいては、遺伝子型と表現型の関係に関する発見が、なぜ、そしてどのようにして一部の個体だけが一般的な疾患を発症するのかという理解を革命的に変えることが期待されています。精神医学では、精神疾患の病原性メカニズムに関する現在の知識が乏しいため、この期待は特に強いです。
いくつかの顕著な例外を除いて、精神医学遺伝学の分野は何十年もの間、精神疾患の遺伝子連鎖と関連を特定し確認するのに苦労してきました。しかし最近、ハイスループットシーケンシングの進歩と、大規模な共同研究による十分なサンプルサイズの組織化により、待望の結果が得られ始めています。2010年以前は、統合失調症のリスクに影響を与える確認された遺伝子変異は発見されていませんでしたが、それ以来、厳密に設計された遺伝子マッピング調査で250以上のリスク変異が特定され、再現されています。アルツハイマー病、自閉症、双極性障害、うつ病を含む他の精神疾患でも同様の成功が見られ始めています。精神疾患に関連する遺伝子のリストが増加していることは、精神疾患の生物学的基盤に関する現在の理解を再定義することを約束しています。遺伝子マッピングの新しい発見は、気分障害や精神病性障害の発症にモノアミン系システムの変化が根本的であると焦点が当てられてきた過去50年間の一般的な理論を支持していません。むしろ、遺伝的発見は、精神疾患の病因が、神経発達、シナプス機能、イオン調節、免疫/炎症、細胞内シグナル伝達を含む、部分的に重複する複雑な細胞プロセスのネットワークに関連していることを示しています。さらに、遺伝子解析は、長らく疑われていたことを確認しています。すなわち、カテゴリ的に定義された診断グループ内に実質的な異質性があり、異なるカテゴリの障害間に有意な遺伝的重複があるということです。疾患の発症に大きな影響を与える1つまたは少数のまれな遺伝的変異を特定することを目的とした伝統的な遺伝学アプローチ(すなわちメンデル遺伝子)とは対照的に、最近の成功の多くは、それぞれが疾患リスクのわずかな増加にしか寄与しない一般的な遺伝子変異の特定に焦点を当てることによって得られています。共通の遺伝子変異とまれな遺伝子変異の相対的な寄与を解明するなど、解決すべき主要な問題がまだありますが、全体として、この分野はついに、精神疾患の病態生理学に関する多くの深い未知が形をなし始めるような、経験的に厳密な基盤を構築し始めています。
遺伝子マッピング研究は、染色体上の位置に基づいて、遺伝性疾患に関与する特定の遺伝子と配列変異を特定することを目的としています。これらの研究は、罹患した個人とその家族を連鎖(連鎖解析)と関連(関連解析)という2つのアプローチで調査することによって行われます(図1.19-1および図1.19-2)。これら2つのアプローチにはそれぞれ明確な利点があり、以下でより詳細に議論されています。簡単に言うと、連鎖法は関連する家族構成員からなるサンプルで実施され、疾患形質に大きな影響を与える遺伝子(高い浸透度)をマッピングするのに適しています。一方、関連法は通常、異系交配集団(非家族構成員)で実施され、疾患に小さな影響を与える遺伝子(低い浸透度)を特定するように設計されています。現在、特定の遺伝子型が特定の遺伝子座で形質を引き起こすために必要かつ十分である形質である、大きな効果を持つメンデル遺伝形質を遺伝的にマッピングするために連鎖法を使用することは簡単です。しかし、精神疾患は単純なメンデル遺伝パターンに従わず、むしろ病因的に複雑な形質の例です。病因の複雑さは、以下の多くの要因による可能性があります。
- 不完全浸透:疾患関連遺伝子型を持つ個体の一部のみで表現型が発現する。
- フェノコピーの存在:遺伝的要因によって引き起こされない疾患の形態。
- 遺伝子座異質性:異なる家族や集団で同じ疾患に異なる遺伝子が関連する場合に発生する。
- 多遺伝子性遺伝:複数の遺伝子における感受性変異が協調して作用する場合にのみ疾患リスクが増加する。
複雑な疾患のマッピングには、研究対象となる表現型の定義、その表現型の遺伝的伝達の証拠を決定するための疫学研究、情報豊富な研究集団の選択、および適切な実験的および統計的アプローチの決定を含むいくつかの構成要素のステップが含まれます。本章では、遺伝的に影響を受ける形質および疾患を特定するために使用される疫学的方法の概要から始め、次に複雑な形質に関連する特定の遺伝子変異をマッピングするために使用される様々なアプローチについて説明します。本章の最終部では、いくつかの主要な精神疾患の遺伝子マッピングにおけるこれまでの進捗状況をレビューします。
疫学
遺伝子疫学アプローチ
特定の遺伝子をマッピングする前に、遺伝子疫学調査は、特定の形質が家族内でどの程度集積するかに関する定量的な証拠を提供し、さらに、そのような集積が形質の病因への遺伝的寄与をどの程度反映しているかを示唆することができます。家族研究は、罹患した個人の親族における疾患の集積を対照サンプルと比較します。これらの研究は、そのような家族性集積に対する遺伝的寄与と環境的寄与を区別しないため、形質の遺伝率に関する間接的な証拠のみを提供します。これらの研究では、罹患した個人の特定の種類の親族における疾患の発生率と、一般集団における疾患の発生率を比較して定義される**家族相対リスク(λ)**を測定することがよくあります。相対リスクが1より大きい場合、遺伝的病因が示唆され、その値の大きさが疾患への遺伝的寄与の推定値を示します。相対リスクは、兄弟姉妹ペア、親子ペア、およびその他の様々な種類の家族関係について計算できます。各関係の種類に対する相対リスクの程度を比較することにより、考えられる伝達様式を評価できます。大うつ病、双極性障害、統合失調症、強迫性障害(OCD)など、多くの主要な精神疾患について複数の家族研究が実施されています。これらの研究は、これらのすべての疾患で家族性集積を一貫して報告していますが、そのような集積の程度は研究間で大きく異なっており、主に表現型の定義と研究サンプルの選定および評価方法の違いを反映しています(詳細については、以下の「表現型」セクションを参照)。
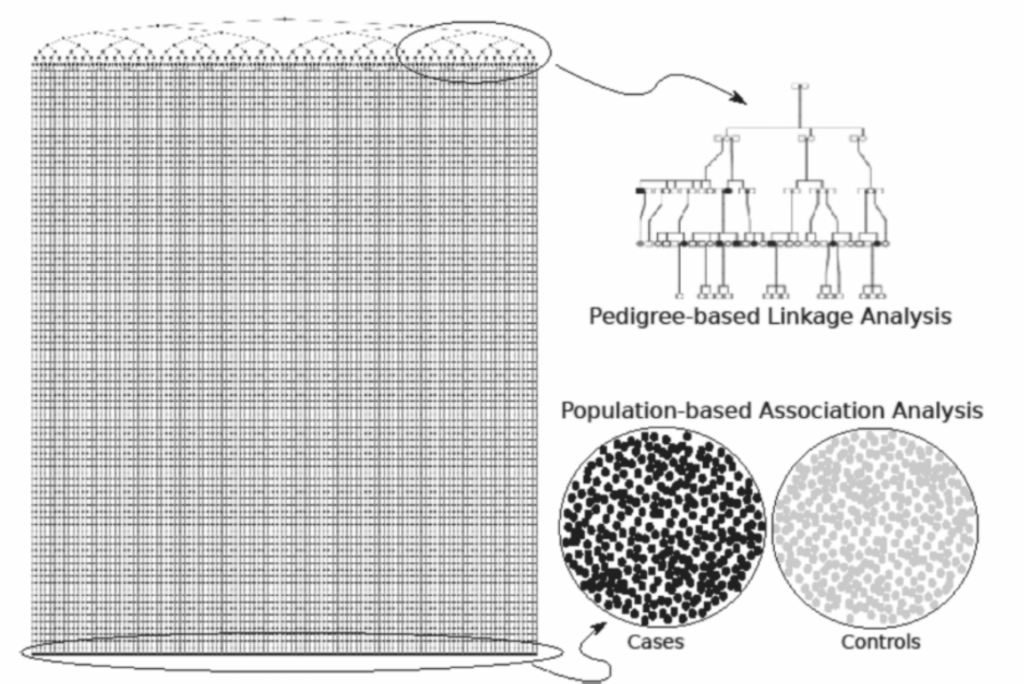
**図1.19-1. 遺伝子をマッピングするための2つの基本的なアプローチ:連鎖(右上)と関連(右下)。**すべての遺伝子マッピングアプローチは、IBD(identical by descent、共通祖先由来同一)で遺伝する遺伝子変異を特定することに基づいています。図の左側は、図の中央上にある創始者と、リスクアレルIBDを遺伝するすべての子孫の模式図を示しています。連鎖解析(右上)は、家族内の複数世代の個人をサンプリングし、既知の場所の遺伝子マーカーと罹患個体における疾患形質の共分離を特定することを目指します。連鎖解析は、遺伝的変異の起源(自然選択によって変異が除去される前)に比較的近い家族構成員における大きな効果を持つ遺伝子を検出するのに最適です。対照的に、関連解析(右下)は、創始者から非常に遠い世代をサンプリングし、対照と比較して罹患個体内の特定の遺伝子変異の濃縮を特定しようとします。何世代にもわたって存続してきた遺伝子変異は、疾患リスクのごくわずかな増加をもたらす傾向があり(一般的に適応度を低下させると予想される)、これは関連研究によって特定された変異の小さな効果量に反映されています。
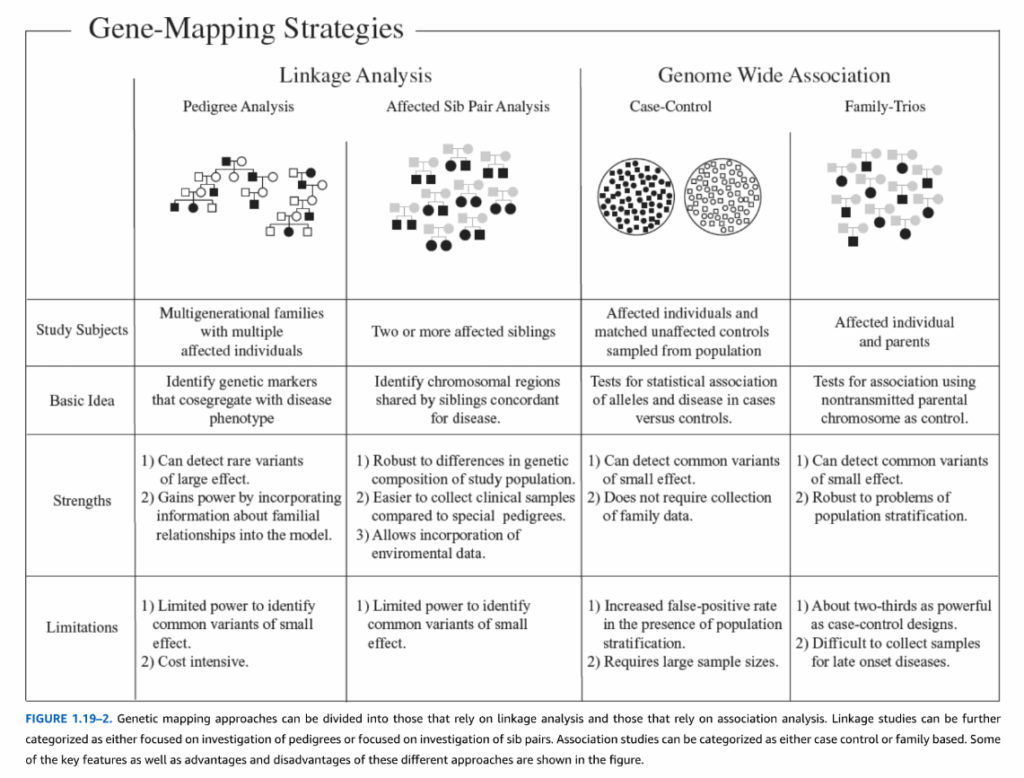
精神疾患の遺伝子マッピング研究アプローチの比較
| 項目 | ペディグリー解析 (Pedigree Analysis) | 罹患同胞ペア解析 (Affected Sib Pair Analysis) | ケースコントロール研究 (Case-Control) | 家族トリオ研究 (Family-Trios) |
| 研究対象者 | 複数の罹患した個人を含む多世代家族 | 2人以上の罹患した兄弟姉妹 | 集団からサンプリングされた罹患個体およびそれにマッチした非罹患対照 | 罹患個体とその両親 |
| 基本的な考え方 | 疾患表現型と共分離する遺伝子マーカーを特定する | 疾患に一致する兄弟姉妹間で共有される染色体領域を特定する | 患者と対照群における対立遺伝子と疾患の統計的関連性を検査する | 伝達されなかった親の染色体を対照として関連性を検査する |
| 長所 | 1) 大規模な効果を持つ稀な変異を検出できる。<br>2) 家族関係の情報をモデルに組み込むことで検出力が向上する。 | 1) 研究集団の遺伝的構成の違いに対して頑健である。<br>2) 特殊な家系図に比べて臨床サンプルの収集が容易である。<br>3) 環境データを組み込むことができる。 | 1) 小規模な効果を持つ一般的な変異を検出できる。<br>2) 家族データの収集が不要である。 | 1) 小規模な効果を持つ一般的な変異を検出できる。<br>2) 集団層別化の問題に対して頑健である。 |
| 短所 | 1) 小規模な効果を持つ一般的な変異を特定する検出力が低い。<br>2) コストがかかる。 | 1) 小規模な効果を持つ一般的な変異を特定する検出力が低い。 | 1) 集団層別化が存在する場合、偽陽性率が増加する。<br>2) 大規模なサンプルサイズが必要である。 | 1) ケースコントロールデザインの約3分の2の検出力しかない。<br>2) 遅発性疾患のサンプル収集が困難である。 |
図1.19-2. 遺伝子マッピングのアプローチは、連鎖解析に依拠するものと関連解析に依拠するものに分けられる。
連鎖研究は、さらに家系図の調査に焦点を当てるものと、同胞ペアの調査に焦点を当てるものに分類できます。関連研究は、ケースコントロール(症例対照)または家族ベースに分類できます。これらの異なるアプローチの主要な特徴、利点、欠点を図に示します。
双生児研究は、一卵性双生児(MZ)と二卵性双生児(DZ)における特定の疾患の一致率(双生児ペアの両方が疾患を持つ割合)を調べます。遺伝的要因のみによって厳密に決定される疾患の場合、一致率はMZ双生児ペア(遺伝物質を100%共有する)では100%であり、DZ双生児ペア(他の兄弟姉妹と血縁関係は変わらない)では、疾患が劣性か優性かによってそれぞれ25%または50%となるはずです。遺伝的要因が疾患の原因に関与するが、疾患の排他的な原因ではない疾患の場合、一致率はMZ双生児の方がDZ双生児よりも高くなるはずです。MZ双生児の一致度が高いほど、形質の遺伝率が高いか、疾患リスクへの遺伝的寄与の証拠が強いことを示します。遺伝的要因が関与しない場合、MZ双生児ペアの環境がDZ双生児ペアの環境と変わらないという単純な仮定の下では、双生児ペア間で一致率に差はないはずです。自閉症、双極性障害、統合失調症などの形質について実施された双生児研究は、一貫して高い遺伝率を示唆しており、したがって、これらの各状態の遺伝子座を遺伝的にマッピングする努力を促進してきました。
しかし、異なる双生児研究は、任意の疾患の遺伝率について異なる推定値を生成する可能性があります。したがって、双生児研究の結果を評価する際には、表現型がどのように確認されたかを精査することが重要です。家族研究と同様に、異なる遺伝率の推定値は、表現型の評価および定義方法の違いに起因する可能性が高いからです。例えば、精神疾患に関する初期の双生児研究は、その表現型を単一の臨床医による非構造化面接に頼ることが多かったのです。対照的に、現代の研究では、一般的に標準化された評価と、専門の臨床医のパネルによる診断資料のレビューが用いられます。同様に、異なる双生児研究間における遺伝率の明らかなばらつきの一部は、ある研究では特定の表現型に対する罹患性の狭い定義を採用しているのに対し、他の研究ではより広い表現型定義を採用している(例:大うつ病(MDD)と診断された双生児が、双極性障害と診断された同胞と表現型的に一致するとみなす)という事実に起因する可能性があります。このようなアプローチの違いにより、これらの研究を形質の変動性に対する遺伝的寄与の概算を提供するものと見なすことが通常は賢明です。それにもかかわらず、そのような推定値でさえ、どの形質がマッピング可能であるかを決定する上で有用です。
遺伝子マッピングの基本概念
導入
特定の表現型の遺伝子疫学研究が、これらの表現型が遺伝性であることを示唆した後、疾患のリスクに寄与する特定の遺伝子変異を特定するために、遺伝子マッピング法が実施されます。遺伝子マッピングの基本的な本質は、染色体上の遺伝子座の順序を特定し、それらの間の距離を決定することです。DNAが遺伝子の分子基盤として発見される前は、マッピング研究は通常、疾患表現型とABO式血液型のような酵素的または生理学的表現型との間の家族における伝達パターンを特定して連鎖を確立しようとしていました。現在のマッピング研究は、既知の場所のDNA多型の広範なセットから始まり、ゲノムを体系的に横断して、未知の疾患遺伝子座の位置を特定します。遺伝子をマッピングするための基本的なアプローチは2つあります:連鎖と関連です(図1.19-1および図1.19-2)。連鎖解析は、家族からの情報を使用して、偶然に予想されるよりも頻繁に世代を超えて一緒に伝達される遺伝子座を特定します。対照的に、関連デザインは家族情報を明示的に使用せず、既知の場所の遺伝子変異と疾患形質の非ランダムな共起を特定することを目指します。連鎖解析は、疾患形質に比較的大きな影響を与える稀な遺伝子変異を特定するのに理想的に適しているのに対し、集団ベースの関連法は、疾患形質に控えめな影響を与える一般的な遺伝子変異を特定するように設計されています。以下のセクションでは、すべてのマッピング法の基礎となる組換えと連鎖の現象から始めて、遺伝子マッピングの基本概念の概要を説明します。
組換えと連鎖
すべての遺伝子マッピング法は、染色体上の位置と遺伝的連鎖の原理に基づいて疾患関連変異を特定することを目指しています。すべての細胞は、各染色体を2コピー(同種体と呼ばれる)持っています。1つは母親から、もう1つは父親から遺伝します。減数分裂中、親の同種体は交差または組換えし、子孫に伝えられるユニークな新しい染色体を作成します。単一の染色体内の連続したDNA鎖上で物理的に互いに近い遺伝子は遺伝的に連鎖しており(一緒に遺伝する傾向がある)、互いに遠い遺伝子または異なる染色体上にある遺伝子は遺伝的に非連鎖です。非連鎖の遺伝子はランダムに組換えを起こします(すなわち、各減数分裂で50%の組換えの可能性があります)。連鎖した遺伝子座は、ランダムな分離によって予想されるよりも頻繁に組換えを起こしにくく、組換えの程度はそれらの間の物理的距離に比例します(すなわち、物理的距離が近いほど、遺伝子座はより頻繁に一緒に伝達されます)。
連鎖の原理は、既知の染色体位置のDNAセグメントであり、バリエーションまたは多型(以下でより詳細に説明)を含む遺伝子マーカーの使用の根底にあります。疾患遺伝子をマッピングする戦略は、罹患した個人によって偶然に予想されるよりも大きく共有される遺伝子マーカーアレルを特定することに基づいています。このような共有は、疾患遺伝子座とマーカー遺伝子座との間の連鎖を反映していると推定されます。すなわち、両方の遺伝子座のアレルが共通の祖先から「同一祖先由来同一」(IBD)で遺伝し、さらに、この連鎖が疾患遺伝子座の染色体上の部位を特定するということです。
2つの遺伝子座間の連鎖の証拠は、それらの間の組換え頻度に依存します。組換え頻度は**組換え率(θ)**によって測定されます。これは、2つの形質または遺伝子座が一緒に伝達される頻度を観察することによって経験的に決定でき、2つの遺伝子座間の遺伝的距離を推定するために使用できます(1%の組換えは遺伝的距離で1センチモルガン[cM]に等しく、平均して約1メガベース[mB]のDNAの物理的距離をカバーします)。組換え率が0.5%または50%は、2つの遺伝子座が連鎖しておらず、独立して分離していることを示しますが、組換え率が低い場合は、遺伝子座が互いに十分近くに位置しており、一緒に伝達される傾向があることを示します。組換え率が低いほど、遺伝子座間の距離は小さくなります。
実際には、経験的に導き出された観測値からオッズ比の対数(LOD)スコアが推定され、特定の遺伝的距離で2つの遺伝子座が連鎖している可能性が決定されます。LODスコアの中心的な統計量はオッズ比であり、これは、特定の組換え率で遺伝子座が連鎖している場合に観測データが得られる可能性を、遺伝子座が連鎖していない場合(θ = 0.5)にデータが得られる可能性で割ることによって計算されます。このオッズ比の対数(底10)がLODスコアです。LODスコアが3.3は、2つの遺伝子座が独立して分離しているのではなく、連鎖している可能性が約1,000対1であることを意味します。LODスコアは、θ = 0(完全に連鎖)からθ = 0.5(非連鎖)までの組換え率の様々な値について得られます。最も大きなLODスコアを与えるθの値は、疾患遺伝子座とマーカー遺伝子座間の組換え率の最良の推定値と見なされます。この組換え率は、その後、2つの遺伝子座間の遺伝子地図距離に変換することができます。実際には、正確なLODスコアを推定するためには、大規模な家族および/または多数の小規模家族を含むサンプルが必要です。
連鎖不平衡 (Linkage Disequilibrium)
連鎖不平衡 (LD) は、家族ベースの連鎖測定に類似した現象ですが、家族ではなく集団における遺伝子座間の遺伝的距離を評価するために使用されます。2つの遺伝子座の対立遺伝子が、それぞれの対立遺伝子頻度から予想されるよりも頻繁に集団内で共起する場合、それらの対立遺伝子はLDにあると言われます。2つの遺伝子座間で強いLDが観察される場合、通常、それら2つの遺伝子座が特定の染色体上で物理的に非常に近い位置にあることを示しており、ある遺伝子座の存在から別の遺伝子座の存在を予測できるため、疾患感受性遺伝子座のマッピングに役立ちます。この予測可能性は重要です。なぜなら、現在の遺伝子マッピング戦略では、推定される1,000万の一般的なヒト多型のサブセットしかサンプリングできないからです。LDの存在により、遺伝子型が決定された多型のサブセットからのデータを使用して、近くの遺伝子座の遺伝子型を推論することができます。LDにあり、単一の単位として遺伝する対立遺伝子のクラスターはハプロタイプと呼ばれます。したがって、LDマッピングは、集団内のハプロタイプを特定することによってゲノム情報を「統合」し、そのハプロタイプを使用して、無関係な個人間のIBD共有(後述の関連法を用いた遺伝子マッピングの中心的な原則)を推論することができます。
LDの範囲を測定する方法はいくつかあります。最も一般的に使用されるLDの尺度の1つはr²で、これは観察されたハプロタイプ確率と期待されるハプロタイプ確率の差の尺度です。r²値が大きいほど、2つの対立遺伝子間の関連性の観察頻度が偶然に予想されるよりも大きいこと、すなわち、対立遺伝子がLDにあることを示します。LD研究は、伝統的に、例えば、連鎖解析によってマッピングされた遺伝子座を絞り込むために、伝統的な家系図解析を補完する形で使用されてきました。しかし、LDベースの関連解析は、特に伝統的な連鎖研究が成功しなかった疾患において、全ゲノムスクリーニングの主要な方法となっています。これらの研究は、伝統的な家族解析に比べて大きな利点があります。罹患した個人が1つまたは少数の家系図からではなく、集団全体から選択されるため、潜在的な被験者の数は集団の規模と疾患の頻度によってのみ制限されます。遺伝的異質性や不完全浸透が要因となる可能性のある疾患では、解析に含めることができる罹患個体の潜在的な数を最大化することが極めて重要です。
遺伝子マーカー
マッピング研究は、その種類に関わらず、遺伝子マーカーの利用可能性に依存します。最も広く使用されているマーカーは、一塩基多型(SNPs)とマイクロサテライトマーカー(単純タンデムリピート[STRs]、または単純配列長多型[SSLPs]とも呼ばれる)です。SSLPsは、2〜4塩基対の長さの反復ヌクレオチドが可変的な数で繰り返される領域です。これらのマーカーは、任意のSTR遺伝子座における反復単位の数が個体間で大きく異なるため、高度に多型性を示します。SNPは、その名の通り、特定のヌクレオチドにおける単一塩基対の変化であり、ゲノムにおける最も一般的な配列変異の形態です。SNPは、ゲノム全体に広く分布しており、ハイスループットかつ自動化された方法で評価できるため、遺伝子マッピング研究に広く使用されています。遺伝子マーカーとして使用が検討されている他の形態の遺伝的変異には、通常1〜30塩基対の範囲の小さな挿入または欠失多型であるインデル(indels)、および欠失または重複のいずれかを指す**コピー数多型(CNVs)**が含まれます。最近のゲノムワイド調査により、CNVsが一般的であり、長さが数塩基対から数百万塩基対に及ぶことが明らかになりました。CNVsは染色体組換えや再配列に寄与し、それによって遺伝的多様性を生み出す上で重要な役割を果たしている可能性があり、また、これらの変異の多くがかなりのサイズであるため、変異を包含する遺伝子または変異に隣接する遺伝子の発現に有意な影響を与える可能性があると仮説が立てられています。
マッピング戦略
疾患感受性に寄与する遺伝子変異は、おおまかに高浸透度と低浸透度に分類できます。高浸透度変異は、定義により、表現型に大きな影響を与え、したがってこれらの変異を特定することは、通常、疾患に寄与する生物学的経路への基本的な洞察を提供します。高浸透度変異を持つ個人は、疾患表現型を発現する可能性が高いため、このような変異は稀であり、家族内で分離し、一般的に家系図ベースの連鎖マッピングアプローチ(図1.19-1および図1.19-2)を用いて最も強力にマッピングされます。対照的に、低浸透度変異は表現型に比較的弱い影響しか与えず、したがって、個々の低浸透度変異を特定することは、少なくとも最初は、比較的少ない新しい生物学的知識しか提供しない可能性があります。しかし、その効果が小さいため、このような変異は集団中で一般的である傾向があり、したがって、それらを特定することは、集団全体の疾患リスクの理解に貢献する可能性があります。これらの変異が家系図内で疾患表現型と強く分離することは期待されておらず、それらを特定する努力は集団サンプルに焦点を当て、表現型への比較的影響が小さいため、大規模なサンプルサイズが必要です。以下のセクションでは、2つの一般的な家族ベースの連鎖戦略(家系図解析と同胞ペア解析)について説明し、次に集団ベースおよび家族ベースの関連解析の概要を説明します。
家系図解析 (Pedigree Analysis)
家系図解析の主な目標は、2つ以上の遺伝子座(すなわち、既知の場所の遺伝子マーカーと未知の疾患遺伝子座)が家系図内で共分離しているかどうかを判断することです。多世代家族で実施される典型的な家系図解析は、1つまたは複数の罹患した家系図で一連のマーカーを用いてゲノムまたはゲノムの一部をスキャンし、各マーカー位置でマーカーと疾患表現型との間のLODスコアを計算し、それによって疾患形質に連鎖した染色体領域を特定することから構成されます。
ハンチントン病などのメンデル遺伝性疾患のマッピングに家系図解析が成功裏に適用された後、多くの研究者が精神疾患遺伝子のマッピングにこの戦略を採用しましたが、成功はせいぜいまちまちでした。1980年代後半から1990年代半ばにかけて、いくつかの家系図ベースの研究がアルツハイマー病、双極性障害、統合失調症の感受性遺伝子座のマッピングを報告しました。アルツハイマー病の3つの遺伝子座に関する連鎖発見は比較的迅速に再現されましたが、双極性障害と統合失調症について報告された発見は最終的に偽陽性であったと判断されました。家系図ベースのアプローチが精神疾患遺伝子座のマッピングに失敗したことについては、様々な異なる説明が提案されていますが、ほとんどの研究者は現在、これらの研究が精神疾患の明らかな病因的複雑性を考慮すると、一般的に著しく検出力が不足していたことを認識しています。
精神医学における家系図解析は、より適切に検出力を持つアプリケーション、すなわち**量的形質遺伝子座(QTLs)**のマッピングへとますます転換しています。QTLsは、疾患診断のようなカテゴリー的形質とは対照的に、連続的に変化する形質の変動に寄与する遺伝子座と定義されます。QTLsは通常、小さな効果を持つ遺伝子座であり、集団における形質の観察された分散の一部にしか寄与しません。現在では、1990年代後半に開発された分析方法を使用することで、脳や行動表現型を含む精神疾患の理解に関連する幅広い量的形質をマッピングするために家系図研究を使用できる可能性があることが一般的に受け入れられています。現在、このような研究がいくつか行われており、通常、家系図の各個人で複数の表現型が評価されています。量的表現型のより詳細な議論は、集団ベースのマッピング戦略の概要の後に提供されます。
同胞ペア解析 (Sib Pair Analysis)
1935年に最初に提案された罹患同胞ペア(ASP)解析は、1990年代に多くの精神疾患を含む複雑な形質の遺伝子マッピングに広く使用されるようになりました。同胞ペア解析は、ある形質について一致した同胞ペアが、ランダムな分離の下で予想される頻度と比較して、ゲノムの特定領域を共有する頻度を調べます。同胞ペア解析は、兄弟姉妹がゲノムの約50%をIBDで共有するという事実に基づいています。したがって、特定の形質に罹患した一連の無関係な同胞ペアが、ゲノムの特定の領域を50%(ランダムな分離条件下で予想される共有割合)を著しく超える頻度で共有する場合、そのゲノム領域は当該形質に連鎖している可能性が高いです。この方法では、兄弟姉妹の遺伝子型が決定され、集団頻度と親の遺伝子型を使用して、各同胞ペアの各サイトにおけるIBDで共有される遺伝子の割合が推定されます。次に、連鎖解析は、各遺伝子座で一致するペアと不一致のペアを比較します。
家系図研究と同様に、ASP研究は、小さな効果を持つ遺伝子よりも大きな効果を持つ遺伝子を特定する検出力が高くなっています。この限界は、罹患した兄弟姉妹での初期連鎖研究後に、追加のマーカーや家族構成員を組み込む2段階設計や、サンプルサイズの増加によって部分的に対処できます。大規模な家系図のすべての構成員を特定し評価するよりも、大規模な罹患同胞セットを特定し評価する方が一般的に労力が少なくて済みます。特に、複数の場所から確認された同胞ペアからのサンプルと表現型データを含むデータリポジトリを活用できる場合はそうです。例えば、米国国立精神衛生研究所(NIMH)は、統合失調症、双極性障害、自閉症、アルツハイマー病に罹患した相当数の同胞ペアのそのようなリポジトリを維持しています。ASP設計の追加の利点は、疫学情報を組み込むことができるため、環境および遺伝子と環境の相互作用を同時に調べることができる点です。
関連研究 (Association Studies)
過去数十年間で、関連研究が、複雑な疾患のリスクの多くを支えていると考えられている比較的小さな影響の遺伝子座をマッピングするための連鎖アプローチよりも強力であるという見方がますます受け入れられてきました。連鎖研究が家族内での遺伝子マーカーと疾患遺伝子座の共分離を見つけようとするのに対し、関連研究は、特定の対立遺伝子が集団内の罹患した個人において予想よりも頻繁に発生するかどうかを調べます。本章で前述したように、関連研究を用いた遺伝子マッピングは、疾患遺伝子を密接に取り囲むマーカーの特定の対立遺伝子がその遺伝子とLDにあるという考えに基づいています。すなわち、これらの対立遺伝子は、IBD(共通祖先由来同一)で遺伝するため、偶然の分離によって予想されるよりも頻繁に罹患した個人に受け継がれます。
関連研究には2つの一般的なアプローチがあります(図1.19-2)。ケースコントロールデザインと、通常トリオ(母親、父親、罹患した子孫)を調査する家族ベースデザインです。ケースコントロール研究では、無関係な罹患個体群とマッチした対照サンプルとの間で対立遺伝子頻度が比較されます。このデザインは、通常、家族ベースのデザインよりも強力です。なぜなら、罹患患者と対照の大規模なサンプルはトリオよりも収集が容易であり、必要な遺伝子型決定の個体数が少ないため安価だからです。ケースコントロールサンプルは、発症年齢が遅い形質(アルツハイマー病など)で、罹患個体の両親が通常利用できない場合に、唯一現実的なデザインである可能性があります。ケースコントロールアプローチの主な欠点は、集団層別化の潜在的な問題です。ケースとコントロールが注意深くマッチングされていない場合、疾患との関連性ではなく集団の違いを反映する対立遺伝子頻度の大きな違いを示す可能性があります。
家族ベースの関連研究は、集団層別化の問題を軽減するように設計されています。このデザインでは、伝達されなかった染色体(親から子に伝達されなかった各染色体のコピー)が対照染色体として使用され、伝達された染色体と伝達されなかった染色体の対立遺伝子頻度の違いが調べられます。これにより、比較グループが定義上、症例グループと遺伝的に類似しているため、層別化の問題が排除されます。ケースコントロール研究よりも集団層別化に対して頑健ですが、家族ベースの研究は、同じ数の罹患個体を使用した場合、約3分の2の検出力しかありません。
過去には、利用可能なSNPが比較的少なかったため、ゲノムワイドな関連研究を行うことは現実的ではありませんでした。そのため、関連研究は、特定の疾患に関連する仮説上の機能に基づいて選択された候補遺伝子の1つまたは少数のマーカーをテストすることに焦点を当てていました。しかし、ゲノム全体に比較的均等に分布する数百万のSNPを特定する国際的な努力と、それらを比較的安価に遺伝子型決定する技術の開発の結果、ゲノムワイド関連(GWA)研究が今や一般的な現実となり、過去10年間で、このような研究は一般的な疾患に寄与する一般的な変異の広範なカタログを生成し始めています。精神疾患における初期のGWA研究では、リスク変異を特定できませんでしたが、最近の研究では非常に大規模なサンプルが使用されており、検出力不足の研究デザインが以前の精神遺伝学的調査の残念な結果の多くを招いたという仮説をさらに裏付けています。
統計的考察
他の生物医学研究分野の科学者は、遺伝学者が連鎖または関連の結果を有意であると見なすために要求する、明らかに高いレベルの統計的証拠にしばしば驚かされます。最も簡単に言えば、この要件は、ゲノムから選択された任意の2つの遺伝子座が互いに連鎖または関連しているという非常に低い期待という観点から考えることができます。任意の2つの特定の遺伝子座が連鎖している可能性(すなわち、連鎖の事前確率)は、ゲノムの遺伝的長さに基づいて約1:50であると予想されます。この低い連鎖の事前確率を補償し、連鎖の事後(または全体的な)確率を約1:20にするために、連鎖に有利な1,000:1の条件付き確率が必要とされます。これは、一般的に受け入れられている有意水準P = .05に対応し、伝統的に受け入れられているLODスコアの閾値3に対応します。これは一般的に許容可能な偽陽性率を提供しますが(図1.19-3)、一部の偽陽性発見は、この閾値さえも超えています。
連鎖研究の許容される有意性閾値は、現在のGWA研究で要求されるはるかに厳格な閾値と比較して比較的低いことに注意することが重要です。より厳格な閾値の理由は、ゲノム内の任意の2つの遺伝子座が互いに関連しているという期待が、それらが連鎖しているという期待よりもさらに低いことです。通常、約10⁻⁷未満のP値が「ゲノムワイドな有意性」を示すと見なされます。任意の2つの遺伝子座が互いに関連している可能性が低いというもう1つの非常に重要な結果は、候補遺伝子研究がゲノムワイド研究で使用されるのと同じ厳格な基準に保持されるべきであるということです。厳格な候補遺伝子研究は、すべての遺伝子座が同じ低い関連の事前確率を持つという不可知論的な仮定を使用し、一部の研究者が特定の障害や形質への仮説上の機能的関連性に基づいて選択された候補遺伝子の変異に割り当てる高い事前確率を無視するでしょう。GWA研究は現在、幅広い複雑な形質について非常に低いP値で関連を再現しており、候補遺伝子関連の大部分(通常、著しく高いP値を報告する)は再現されていません。したがって、ゲノムワイドな有意水準が、定義された候補遺伝子セット内の少数の遺伝子変異に限定される場合でも、特定の形質のすべての初期関連研究に適切に適用されることはますます明らかになっています。遺伝子座と疾患または行動形質との間の関連性について、より厳密に確認された生物学的データが得られるにつれて、将来の関連研究は厳格な不可知論的基準を緩和できるようになるでしょう。例えば、ゲノムワイドマッピング研究が、厳格な閾値を使用して遺伝子座と神経症のような形質との関連性を特定した場合、同じ遺伝子座と不安障害の診断のような類似の形質の将来の研究は、より厳格でない閾値でも意味があると見なされるでしょう。
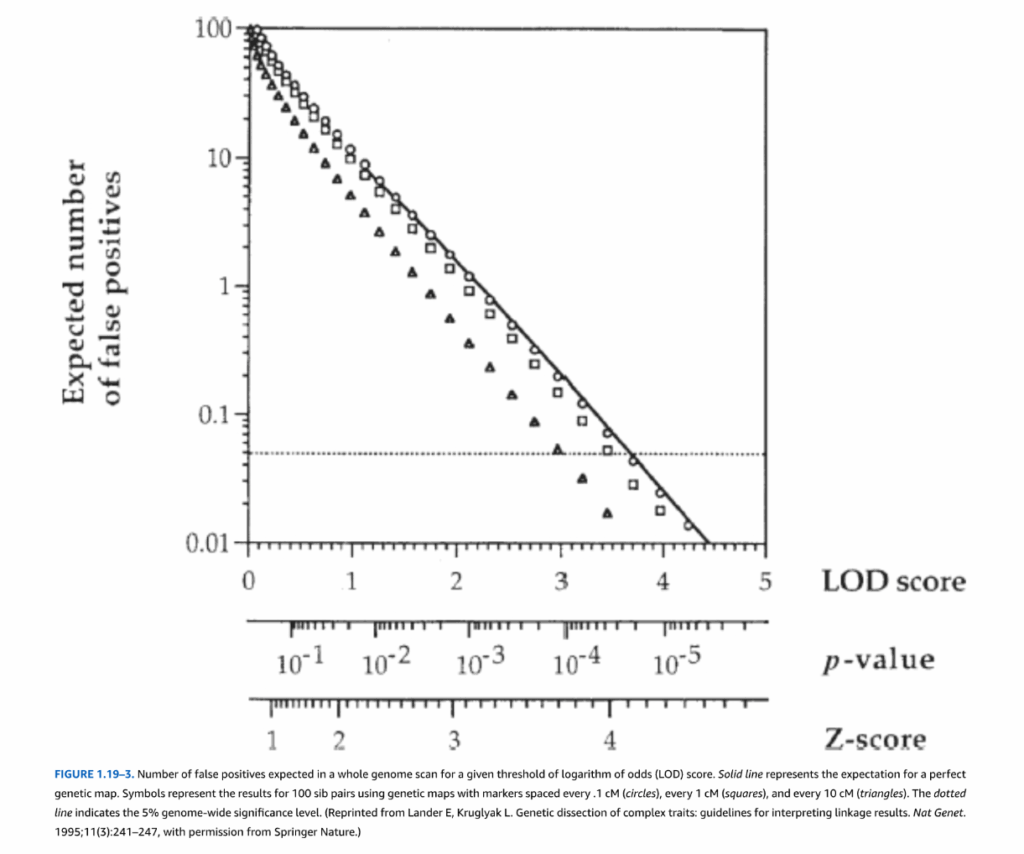
図1.19-3. 特定のオッズ比の対数(LOD)スコア閾値における全ゲノムスキャンで予想される偽陽性の数
実線は、完璧な遺伝子地図における期待値を示します。記号は、マーカーが0.1cM間隔(円)、1cM間隔(四角)、10cM間隔(三角形)で配置された遺伝子地図を使用した100組の同胞ペアの結果を示します。点線は5%のゲノムワイド有意水準を示します。(Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting linkage results. Nat Genet. 1995;11(3):241-247 より、Springer Natureの許可を得て転載。)
表現型
精神疾患の遺伝子マッピング研究で一般的に期待外れな結果が出ていることは、そのような研究における表現型の定義と評価の問題にますます注目が集まっていることを示しています。現在までのほとんどの精神医学マッピング研究は、診断と統計マニュアル(DSM)分類スキームに例示されるような、カテゴリ的な疾患診断に依拠してきました。このアプローチへの批判は、2つの議論に基づいています。第一に、精神疾患の診断は主観的な臨床評価に依存しており、これは特定の疾患に確実に罹患していると見なせる個人を特定することの難しさを強調する事実です。第二に、精神疾患の診断が曖昧さなく確立できる場合でも、精神医学的分類に用いられるメニューベースのシステムは、特定の障害に罹患した任意の2人の個人が、ほとんど重複しない症状のセットを示す可能性があり、これはおそらく異なる病因を反映していると考えられます。
表現型を診断ベースのアプローチで特定することが、精神医学的表現型の遺伝子マッピングにおける主要な障害の1つであるという懸念から、集団内で連続的な変動を示すことが知られている遺伝性形質のマッピングに大きな関心が寄せられています。精神疾患に関連すると仮説されている連続的な測定値には、生化学的測定値(例:血清またはCSF中の神経伝達物質代謝産物またはホルモンのレベル)、認知測定値、パーソナリティ評価、構造的または機能的な脳画像、誘発電位への応答などの生物物理学的マーカー、または遺伝子発現プロファイルなどの分子アッセイが含まれます。カテゴリカルおよび連続的な表現型決定戦略の主な特徴は、図1.19-4および図1.19-5に示されており、それぞれについて以下でより詳細に議論します。
