
Fig 1.19-4
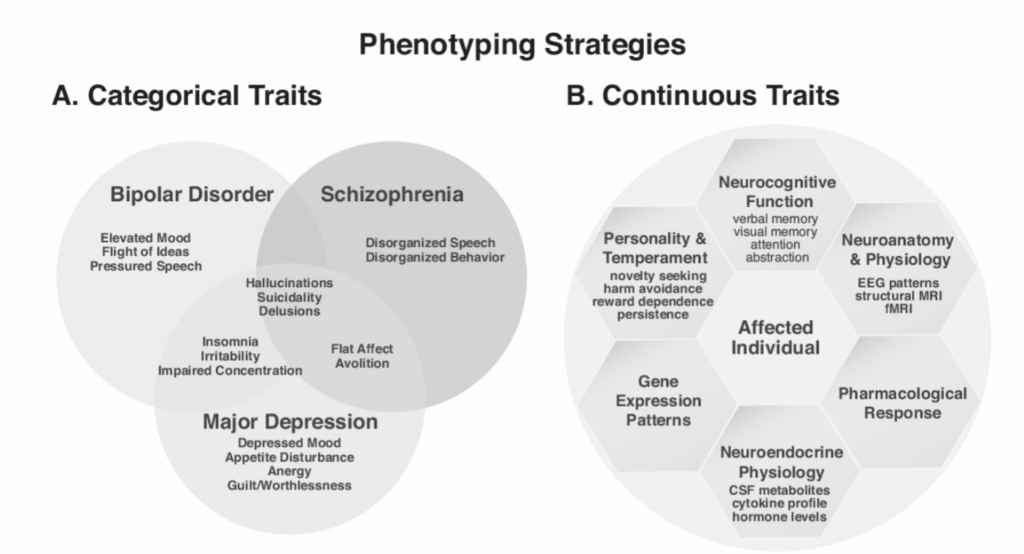
図1.19-4。精神医学的表現型を概念化するための2つの代替的枠組み。
A:診断・統計マニュアル(DSM)によって概念化されるカテゴリー的特性は、精神疾患への「メニュー方式」のアプローチを表す。個人は兆候と症状のチェックリストに基づいて評価され、それを用いて特定の診断に従って「罹患している」と分類される。特定のDSM診断を持つ個人のサンプルにおいて、全ての症状が存在するわけではなく、これらの症状の多くは、このベン図で示されるように、診断の境界を越えて発生する。したがって、DSMの表現型は、病因的に異質なカテゴリーを表している可能性が高い。
B:あるいは、連続的特性モデルでは、「罹患状態」は、個人が精神病理と相関し、したがって疾患の根底にあると仮定される一連の連続的尺度(六角形に示される6つの異なるタイプの尺度の例で示される)において極端な値を示すという期待の観点から概念化することができる。このような尺度はまた、ベン図に示されるような、カテゴリー的表現型の特定の構成要素と関連している場合もある。遺伝子マッピング研究の表現型として連続的尺度を使用することの正当性は、それらがカテゴリー的表現型と比較して、病因的により単純で、より信頼性高く評価されると考えられることである。さらに、そのような特性をマッピングすることは、研究集団の全てのメンバー(罹患者および非罹患者の両方)からの情報を組み合わせるため、検出力を大幅に高める。
カテゴリー的表現型
精神医学で最も一般的に使用されるカテゴリー的表現型は、DSM診断である。一部の研究では単一のDSM診断に焦点を当てるが、他の研究ではさまざまな異なる診断を持つ個人が含まれる。後者のアプローチは、気分障害など、単一の疾患スペクトラムを表すと仮定される障害に典型的に用いられる。カテゴリー的アプローチを用いる場合、被験者をできる限り曖昧さなく分類できることが重要である。この目標を達成するために、いくつかの戦略が用いられる。最初の戦略は、当該研究に適した診断基準を決定し、これらの基準を研究対象の個人にどのように適用するかを決定することを含む。潜在的な研究対象者を特定し評価するために用いられる手順を標準化する一つの方法は、診断プロセスにおいて経験豊富な臨床医のみを用い、使用される(評価)ツールと診断基準の実施について彼らを訓練することである。さらに、「最良推定」手続きやコンセンサス診断が頻繁に用いられる。最良推定プロセスでは、医療記録、面接、ビデオテープを含む、利用可能なあらゆる情報を用いて診断に至る。コンセンサス診断では、2人以上の診断者が独立して資料をレビューし、各個人について診断を下す。その後、診断が比較され、診断の一致が得られない個人は、研究に「罹患者」として組み入れられない。
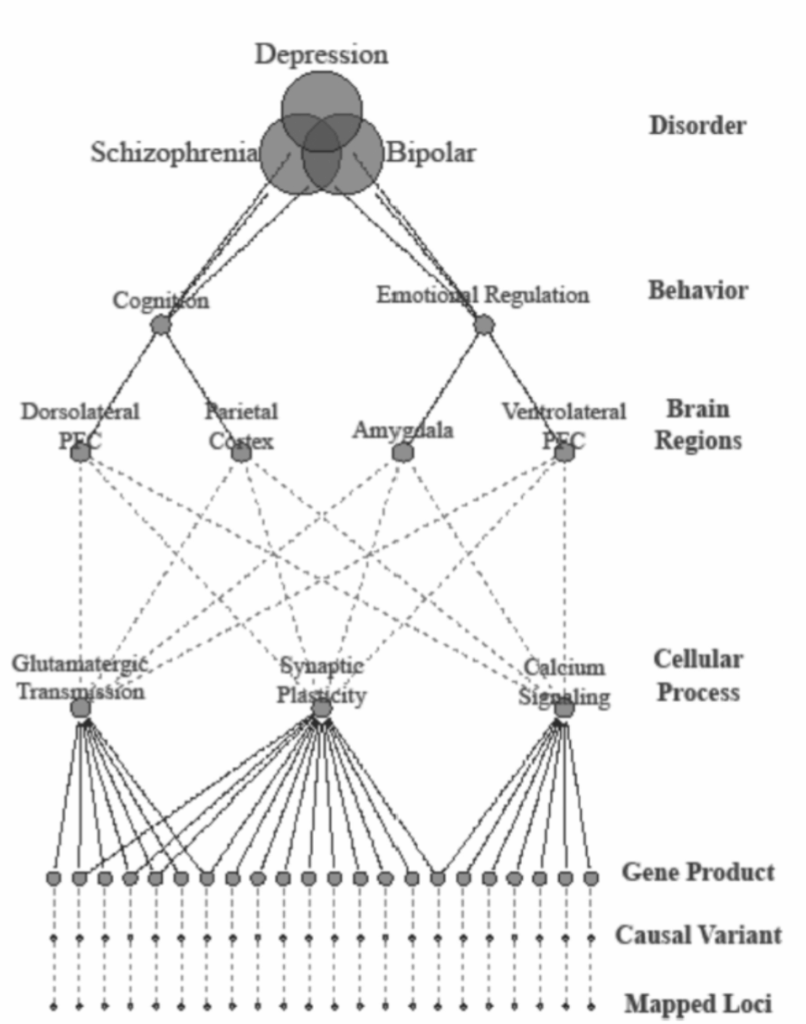
図1.19-5。エンドフェノタイプ(中間表現型としても知られる)戦略は、精神疾患を引き起こす生物学的経路を特徴付けるために、複数の生理学的レベルから得られた定量的尺度に関連する遺伝子をマッピングすることを目的とする。二分法的疾患カテゴリーに関連する遺伝子をマッピングする従来の研究とは対照的に、エンドフェノタイプ法は診断カテゴリーを横断して実施することができ、精神疾患に寄与する生物学的システムの差異や共通性を特定することができる。中間表現型は診断カテゴリーと比較して「遺伝子により近い」ため、遺伝子マッピング研究においてより高い検出力を持つと期待される。
適切に設計された研究は、研究対象とする罹患者のサンプルを選択するために、疾患の遺伝疫学に関する利用可能な全ての情報を活用する。多くの場合、一部の家系では疾患が単純なメンデル様式で遺伝しているように見えるが、他の家系や集団では遺伝様式がそれほど明確ではない。表現型に複数の遺伝子が寄与している可能性が高い疾患においては、主要な遺伝子座が存在する可能性のある研究サンプルから始めるのが理にかなっている。疾患表現型を再定義することは、そのような集団や家系を特定することによって、マッピングプロセスをしばしば単純化することができる。例えば、アルツハイマー病の遺伝的欠陥の探索において、研究集団を発症年齢が早い(65歳以前)個人に限定することで、プロセスは著しく進展した。この早期発症という特性は、常染色体優性形式で分離した。表現型を再定義する他の方法には、民族的背景、治療反応、症状の重症度、または合併疾患の存在などの要因に焦点を当てることが含まれる。
上記で議論されたアプローチを用いて表現型を狭めることは、複雑な疾患における遺伝的欠陥を発見する可能性を高めるかもしれないが、利用可能な罹患者数を制限することにより、研究の検出力を大幅に低下させる可能性もある。この理由から、一部の疾患については、表現型を広げることが適切な戦略であると主張されてきた。その提案は、一部の複雑な疾患では、関心のある表現型がスペクトラムの極端な端を表している可能性があり、遺伝子をマッピングするのに十分な検出力を得るためには、スペクトラム内の他の表現型も含まなければならないというものである。例えば、双極性障害のマッピング研究には、双極性障害と診断された個人だけでなく、大うつ病性障害(MDD)の罹患者も含まれる可能性がある。
疾患表現型を狭めるアプローチと疾患表現型を広げるアプローチは、相互に排他的であるように見えるかもしれないが、複雑な疾患を研究している多くのグループは、両方のアプローチを研究デザインに組み込んでいる。これを行う一つの方法は、狭い診断カテゴリーから広い診断カテゴリーに至る層別化された診断カテゴリーを作成し、これらの各スキーマの下で遺伝的連鎖を検証することである。一部の研究者は、スペクトラムの一部である複雑な疾患の場合、この戦略は偽陰性、すなわち誤特定のために既存の連鎖を見逃す率を低下させると主張する。他の研究者は、複数のモデルを使用し、最も高いスコアを与えるものを選択することは、偽陽性、すなわち連鎖が存在しない領域を連鎖として特定する率を大幅に増加させると主張する。複数の診断カテゴリーを使用する際に明確に存在する一つの問題は、より多くのモデルが使用されるにつれて(したがって、より多くの統計的検定が実行されるにつれて)、結果を有意と見なすためにますます厳密な証拠レベルが必要とされることである。
カテゴリー的表現型は依然として精神医学的遺伝研究の主流であるが、遺伝研究のための表現型判定の基礎としてのDSM疾病分類学の限界は明らかになりつつある。遺伝学的調査は、1つ以上のDSM診断カテゴリーの構成要素である可能性のある特性にますます焦点を合わせている。例えば、広義の精神病への遺伝的感受性が、重症双極性障害と統合失調症の両方に寄与するという証拠が増えており、そのような感受性の根底にある遺伝子を特定し、さらには精神疾患と非精神疾患との間の病因的関係の可能性を探るために、多くの研究的アプローチが採用されている。例えば、バイオインフォマティクスモデルが医療記録データベースを調査するために採用され、精神疾患、神経疾患、自己免疫疾患、および感染症の多様なリスト間で広範なペアワイズ相関が発見されている。最終的には、そのようなモデルフィッティング実験の結果が、複数の疾患への感受性に寄与する対立遺伝子を探索できる、より強力な連鎖研究および関連研究を設計するためのフレームワークを提供するかもしれない。
連続表現型
カテゴリー診断の遺伝的マッピングにおける困難さから、神経行動遺伝学者は、特定の精神医学的診断の根底にあると考えられ、遺伝的マッピングがより単純である可能性のある量的形質の研究にますます焦点を当てています。エンドフェノタイプまたは中間表現型と呼ばれるこのような代替表現型をマッピングする努力の論理的根拠は、そのような努力を通じて同定された遺伝子が、特定の障害または重複する病因経路を持つ可能性のある複数の障害を理解する上で適切な生物学的経路に関する手がかりを提供する可能性があるということです(図1.19-5)。いくつかの特徴が、有用なエンドフェノタイプを特徴づけます。第一に、それらは状態に依存しないべきです。つまり、疾患の経過や投薬治療の関数として変動するべきではなく、十分な再検査安定性を示すべきです。第二に、それらは遺伝性であるべきです。つまり、遺伝的要因が個体群内の形質の変動性の実質的な割合を担っているという証拠があるべきです。第三に、エンドフェノタイプは調査中の疾患と相関しているべきです。つまり、形質測定の異なる値が、関連のない対照被験者と比較して患者で観察されるべきです。脳の構造と機能の尺度は、現在、精神医学的障害のエンドフェノタイプとして調査されています。例えば、脳形態計測のいくつかの特徴(磁気共鳴画像法[MRI]によって評価される)は、総脳容積、小脳容積、灰白質および白質密度、扁桃体および海馬容積、ならびに領域皮質容積を含め、非常に遺伝性が高い(60%から95%の範囲)。いくつかの研究は、統合失調症や双極性障害などの障害と臨床サンプルで相関している脳の構造的特徴が、罹患した個人の親族においても異常であることを示しています。精神医学的障害の候補エンドフェノタイプとして採用されている脳活動の生理学的尺度には、脳波(EEG)パターンが含まれます。脳の測定に加えて、精神医学的障害に関連する行動特性は、神経認知、衝動性、感情調節、および気質(すなわち、神経症傾向)を含め、遺伝子マッピング研究の活発な分野です。
動物モデル
カテゴリー表現型とは対照的に、エンドフェノタイプは、動物モデルで評価できる表現型により直接的に関連付けることができます。概日リズムに影響を与える遺伝的変異の研究は良い例を提供します。概日リズムの変動は、気分障害の重要な特徴として長い間認識されており、活動パターンの定量的評価がそのような障害のエンドフェノタイプとして提案されています。動物モデルにおける多数の研究は、遺伝的に制御された生物時計が概日活動を決定し、時計遺伝子の変異が細菌からヒトまでのそのような活動の変異と関連していることを実証しています。1970年代初頭に始まったショウジョウバエにおける遺伝的マッピングの努力は、periodに始まり、少なくとも7つの「時計遺伝子」の同定をもたらしました。その後の研究は、これらの遺伝子のいくつかの相同体が哺乳類の概日リズムの調節に不可欠な役割を果たしていることを示しました。マウスにおける遺伝的マッピング研究もまた、1990年代初頭のclockの発見と特性評価に始まり、以前は未知であった概日リズム遺伝子を同定しました。これらの遺伝的発見は、哺乳類の概日リズムの制御に関与する細胞ネットワークと神経生理学的回路を明らかにしただけでなく、双極性障害などの精神医学的症候群の病態生物学に光を当てる可能性のある動物モデルも生み出しました。例えば、clockに標的変異を持つマウスは、活動亢進や睡眠減少などの異常な活動パターンを示し、これらはリチウムの投与によって明らかに修飾されます。
特定疾患の遺伝的解明における進展
全体として、精神医学的疾患の感受性遺伝子の同定における進展は、非精神医学的疾患で観察されたものと比較して遅れています。本章の最終セクションでは、いくつかの特定の精神医学的疾患の遺伝的基盤の同定においてなされてきた進展を概観します。アルツハイマー病は、複雑な神経行動学的障害への遺伝子マッピング戦略の最も成功した応用例であり、この疾患に関するセクションは、遺伝的連鎖研究が複雑な形質の病因の理解にどのように貢献するかの例を提供します。自閉症の遺伝子マッピング研究は、比較的単純な遺伝パターンを持つ症候群の遺伝的調査の例を提供し、これらの研究がより複雑な自閉症スペクトラム障害(ASD)の調査の出発点を提供したことを示しています。最後に、統合失調症、双極性障害、および大うつ病のリスク変異体を同定する最近の進展は、これらの複雑な疾患の病因の現在の理解を革命的に変え始めている刺激的な方向性を示すために使用されます。
アルツハイマー病
アルツハイマー病は、神経精神医学的疾患の複雑な生物学を解明するための遺伝学の力を示す優れた例です。アルツハイマー病は、記憶および知的機能の進行性の障害を特徴とする、明確に定義された認知症の一形態です。臨床的兆候および症状は、特徴的ではありますが、アルツハイマー病に限定されるものではなく、他のいくつかのタイプの認知症でも見られます。アルツハイマー病には、いくつかの特徴的な組織病理学的特徴があります。老人斑(ジストロフィー性神経突起に囲まれたβアミロイド線維のコアからなる)、タウリッチな神経原線維変化、および脳実質および関連血管におけるコンゴフィリック血管症の存在は、アルツハイマー病に特有のものです。
アルツハイマー病の発症年齢は変動することが指摘されており、早ければ35歳から遅ければ95歳までの範囲です。家族歴は、年齢に次いでこの疾患の2番目に大きな危険因子です。MZ双生児ペアにおけるアルツハイマー病の一致率は約50%であり、疾患リスクに対する中程度に強い遺伝的寄与を示しています。現在、広範囲の遺伝学的研究から、アルツハイマー病は2つの広範なカテゴリーに分類できることが明らかになっています。家族性形態は、アルツハイマー病症例の約5%を占め、早期発症と高い浸透率を伴う常染色体優性遺伝を特徴とします。そして散発性形態では、遺伝的寄与は他の一般的な神経精神医学的疾患を特徴付けるものと同様であると仮定されています。
家族性アルツハイマー病の遺伝的基盤の探索は、伝統的な連鎖研究から始まりました。まず、ヒトの21番染色体上の候補遺伝子座の調査により、この領域からのマーカーに対して以前に有意な連鎖が観察されていた少数の家族において、APP遺伝子の変異が同定されました。APPは、アミロイド前駆体タンパク質をコードしており、これは細胞表面および膜貫通前駆体タンパク質であり、切断されて様々な機能を持つ複数のペプチドを形成します。ペプチドの一部は分泌され、転写活性化を促進するように機能する一方、他のペプチド産物はβアミロイド線維の基礎を形成します。異なるAPP変異を持つトランスジェニックマウスが作成され、βアミロイド沈着および老人斑を産生し、シナプス喪失、アストロサイトーシス、およびミクログリオーシスを示すことが示されており、これらはすべてアルツハイマー病の病理の一部です。β-APPをコードする遺伝子の変異はすべて、より長いβアミロイド断片(Aβ42)の細胞外濃度の増加につながります。APPに変異を持つトランスジェニックマウスのほとんどの系統は、行動変化の速度の増加およびいくつかの記憶課題における障害を示し、とりわけ物体認識記憶および作業記憶の機能不全を示しています。これらの知見は、βアミロイド遺伝子の変異が実際にアルツハイマー病の組織病理学的要素の少なくとも一部に関与しているという顕著な証拠を表しています。
上記の知見が報告されている間にも、βアミロイド遺伝子の変異がアルツハイマー病の病因と病理を完全に説明できないことは明らかでした。少なくとも、ほとんどの早期発症アルツハイマー病家族において21番染色体への連鎖が除外されたことが示されたためです。さらに、ほとんどの異なるβアミロイドトランスジェニックマウスでは神経原線維変化は観察されません。早期発症アルツハイマー病家族のゲノムワイド連鎖解析を用いたアルツハイマー病の遺伝的基盤のその後の探索は、さらに2つのアルツハイマー病感受性遺伝子の同定をもたらしました。染色体14q24.3上のプレセニリン-1(PS-1)と染色体1q上のプレセニリン-2(PS-2)です。PS-1とPS-2は、少なくとも7つの膜貫通ドメインを持つ膜貫通タンパク質です。プレセニリンは、APPのγセクレターゼ切断を実行してβアミロイドを生成するアスパラギン酸プロテアーゼです。APPの変異と同様に、プレセニリンの遺伝的変異は、より長いβアミロイド断片の発現の増加を引き起こします。
これらの知見は、家族ベースの連鎖解析を使用することの強みの1つを強調しています。家系ベースの研究は、重要な生物学的プロセスにおいて重要な役割を果たす高浸透性の疾患遺伝子を同定するのに特に適しています。APPおよびプレセニリンの変異はまれですが、これらの遺伝子の異なる変異は異なる創始者で発生しています。現在までに、PS-1で185の変異、PS-2で13の変異、APPで24の変異が同定されています。これらの変異のほぼすべてが、常染色体優性かつ完全浸透性の様式で伝達されます。同じ遺伝子内の異なる変異の収束は、これらの遺伝子の疾患への関与を明確に確認します。APPおよびプレセニリンの変異はまれですが、発現タンパク質の生物学の研究は、認知症の病態生理学に関する重要な洞察を提供しています。これらの高浸透性変異は重要な生物学的機能を解明するため、治療的介入を設計するための確固たる基盤も提供します。例えば、病原性アミロイドに対する免疫原性応答を誘導するように設計されたβアミロイド「ワクチン」は、現在、進行した臨床試験段階にあります。さらに、プレセニリン遺伝子産物の機能不全を標的とするために、γセクレターゼモジュレーターが開発されています。コリン作動性およびグルタミン酸作動性システムを非特異的に標的とするアルツハイマー病の現在の精神薬理学的治療とは異なり、βアミロイドワクチンおよびγセクレターゼモジュレーターは、免疫応答を生成するか、APPの処理を変化させることによってアルツハイマー病の原因を特異的に治療し、実際に老人斑の沈着を予防または逆転させる可能性があります。
散発性および遅発性アルツハイマー病
APP、PS-1、またはPS-2の変異は、早期発症アルツハイマー病の家族性症例の大部分に存在するが、散発性または家族性遅発性アルツハイマー病を説明するものではない。このため、研究者らは、遅発性アルツハイマー病を持つ多数の小規模家族における連鎖の証拠を探すために、他のアプローチに目を向けた。1991年、遅発性アルツハイマー病家族における36のマーカーを用いた連鎖研究の結果は、19番染色体の長腕上の感受性遺伝子の証拠を提供した。1993年、関連研究により、アポリポタンパク質E遺伝子のe4対立遺伝子が遅発性アルツハイマー病と強く関連しており、この関連が以前に観察された19番染色体上の連鎖シグナルにほぼ確実に関与していることが明らかになった。この遺伝子には、e2、e3、およびe4の3つの既知の対立遺伝子がある。これらの3つの対立遺伝子は、残基112および158のアミノ酸の組み合わせによって区別される。e2:Cys112/Cys158、e3:Cys112/Arg158、およびe4:Arg112/Arg158。ほとんどの集団では、e3対立遺伝子が最も一般的である。しかし、家族性遅発性アルツハイマー病では、e4の発生率は約50%であり、散発性遅発性アルツハイマー病では40%であり、正常対照の約16%と比較される。e4の単一コピーの遺伝は、疾患のリスクを4倍に増加させ、対立遺伝子の2つのコピーの遺伝はリスクを10倍に増加させる。疫学研究は、遅発性アルツハイマー病症例の30%から60%が少なくとも1つのapoE-e4対立遺伝子を持っていることを示唆している。e4遺伝子型は、アフリカ系の集団と比較して、ヨーロッパおよびアジア系の集団においてアルツハイマー病のより重要な危険因子であるように思われる。apoE遺伝子は、脂質の代謝および輸送において十分に特徴付けられた役割を果たしており、脳からのβアミロイドのクリアランスにおいて役割を果たすと考えられている。全体として、apoE-e4とアルツハイマー病との関連は、一般的なヒト疾患についてこれまでに同定されたおそらく最も強い関連であり続けている。 apoE-e4が遅発性アルツハイマー病の感受性対立遺伝子として確立されたことにより、疾患リスクを修飾するためにapoE-e4と相互作用する可能性のある追加の対立遺伝子の探索が行われるようになった。2007年、研究者らは、組織学的に確認された症例および対照においてGWA戦略を用い、apoE-e4キャリア(ただし、e4キャリアではないアルツハイマー病患者ではない)における追加のリスク対立遺伝子としてGAB2(GRB関連結合タンパク質2)を同定した。初期の研究では、apoE-e4とGAB2の両方のリスク対立遺伝子のキャリアは、どちらのリスク対立遺伝子も持たない個人よりもアルツハイマー病のリスクがほぼ25倍高いことが示唆されている。GAB2は、APPおよびプレセニリンと物理的に相互作用するGRB2(成長因子受容体結合タンパク質2)との相互作用を介してβアミロイド産生を調節するように見える。 GAB2を同定した最初のGWA研究以来、十数件以上の追加のGWA研究があり、それらは合わせて40以上の確認されたアルツハイマー病リスク遺伝子を同定した。APPおよびプレセニリンとは異なり、これらの遺伝子は効果量が小さく(オッズ比<1.2)、一般集団では比較的よく見られる。APPおよびプレセニリンの生物学の現在の理解と比較して、病態生理学におけるそれらの役割はあまり明確ではないが、GWA研究で同定された遺伝子の既知の機能の分析は、免疫、脂質代謝、タウ結合タンパク質、およびアミロイド前駆体タンパク質代謝の関与を示している。注目すべきことに、集団ベースのGWA研究は、APPおよびプレセニリン以外のβアミロイド処理に関連する遺伝子を同定しており、疾患の家族性および散発性の両方の形態における共通の経路への収束を示している。例えば、ATXN1遺伝子産物はγセクレターゼレベルを調節し、APPの切断に影響を与え、ATXN1のマウスノックアウトモデルは生後6ヶ月までに認知障害を示す。GWA研究によって同定された他の遺伝子は、免疫反応に必要な細胞相互作用を調節する因子(例えば、CD33)や炎症プロセス(例えば、CR1、補体分子C3bの受容体)を含む、免疫および炎症経路に影響を与えることによってアルツハイマー病のリスクに影響を与えるように見える。これらの免疫/炎症プロセスは、βアミロイドに対する神経毒性応答を媒介すると仮定されている。 アルツハイマー病のGWA研究の結果が継続するにつれて、リスクに関連する遺伝子と並んで、保護および回復力に関連する遺伝子が出現してきた。無症候性アルツハイマー病としても知られるアルツハイマー病への回復力は、疾患からの保護を与える生物学的経路を同定する可能性があるため、大きな関心を集めている。認知的に正常な高齢者の70%もがアルツハイマー病の組織病理学的変化の明確な証拠を示し、認知的に正常な高齢者の最大30%が疾患の完全な剖検確認された神経病理学的基準を満たしている。脳の病理の明確な証拠にもかかわらず保護を与える遺伝的および環境的要因の特性評価は、疾患プロセスの影響を軽減するための予防または治療戦略に有望性をもたらす。例えば、βセクレターゼ切断部位の近くにあるAPP遺伝子内の最近同定されたミスセンス変異は、疾患に対して保護的であり、認知機能低下を遅らせることが示された。in vitro研究では、この変異により、野生型細胞と比較して、病理学的Aβのその後の凝集が50%減少する、最適ではないβセクレターゼ切断が生じることが示された。このような研究は、最終的に疾患の完全な顕在化をもたらすプロセスを予防または遅延させることを目標として介入するための潜在的な経路を提供する。
要約
アルツハイマー研究の分野における進歩は著しい勢いを達成しており、現在ではこの疾患の病因に関与する40以上の遺伝子が存在する。アルツハイマー病のまれな家族性形態の連鎖研究は、アルツハイマー病の病因の理解およびCNS内の広範囲の細胞プロセスの基本的理解に深遠な影響を与えた高浸透性変異体の発見につながった。関連研究は、集団レベルでの疾患リスクへの遺伝的寄与の多くを共に説明する低浸透性変異体を明確に同定した。総合すると、リスク遺伝子はいくつかの基本的な生物学的経路に収束する:βアミロイドの処理、脂質代謝、免疫、および炎症。リスク対立遺伝子の同定に加えて、遺伝子マッピング研究はまた、疾患からの保護を与える遺伝的変異体を同定した。リスクおよび保護変異体の同定は、2種類の医学的ブレークスルーの可能性を提供した。これらの研究を通じて同定された分子経路を標的とする新しい治療法が開発中である。さらに、アルツハイマー病の一般的な形態の遺伝的リスクの新たな状況は、高リスクの個人に予防および早期介入戦略を集中させることが間もなく可能になるかもしれないことを示唆している。
自閉症
自閉症は、1943年にレオ・カナーによって最初に「自己への閉じこもり」として記述された重度の神経発達障害であり、3つの主要な特徴によって特徴付けられる:言語およびコミュニケーションの障害、異常または障害のある社会的相互作用、ならびに制限された反復的で常同的な行動パターン。自閉症の病因の理解はゆっくりと進んできたが、現在では、特定の細胞および分子的神経発達経路の変化がその病因において重要であるという説得力のある証拠がある。自閉症およびASDのリスクに対する遺伝的寄与については、特に強力な証拠がある。自閉症および/またはASDの同胞再発リスクは2%から6%の間である。人口有病率が約2,000人に1人(0.04%)であることを考えると、これは、自閉症の個人の同胞が一般集団の人よりも自閉症を発症する可能性が約50倍から100倍高いことを意味する。自閉症の双生児研究は、非常に高い遺伝率(MZ双生児の一致率80%から92%によって実証される)を示すだけでなく、これらの障害の遺伝的複雑性も実証しており、DZ双生児の一致率が1%から10%であることは、高度に多遺伝子性の遺伝形式を示唆している。 大規模な染色体異常(CNV)が自閉症において重要な役割を果たす可能性に関心が高まっている。初期の研究では、対照群の1%から3%と比較して、自閉症の個人では5%から10%の率でCNVの頻度の増加が見られた。その後の研究では、最初に見つかった対照群と比較したCNVの高い発生率を再現できなかった。しかし、最近の研究では、CNVの特定の場所と機能的結果が、異常の相対的な数よりも重要であることが示唆されている。2つの大規模な研究は、ゲノム内でランダムに分布しているのではなく、むしろ遺伝子座のサブセット内に集中している異種のCNVのコレクションを同定した。これらの遺伝子座の機能分析は、それらがシナプス形成、神経細胞移動、および軸索ターゲティングに関与していることを示した。これらの機能的知見は、十分に特徴付けられた非CNV関連異常の調査(以下で詳細に説明)とうまく一致しており、散発性または遺伝性のCNVがこの障害の一部の症例にとって重要な病因因子であることを確信させる。全体として、大規模な集団研究では、CNVが症候性自閉症症例の5%から8%を占めると推定されている。 自閉症における新生突然変異の高い発生率が実証される以前から、疫学研究はこの疾患の遺伝的基盤が複雑である可能性を強く示唆していた。例えば、自閉症のプロバンドの第一度近親者における自閉症のリスクは高いが、そのようなプロバンドの第二度および第三度近親者では大幅な減少があり、複数の遺伝的変異体が相互作用してこの症候群への感受性を高める必要があることを示唆している。自閉症の分離分析もまた、それが効果の小さい複数の遺伝的変異体の作用を反映する異種性の障害であるという仮説を支持している。自閉症の遺伝学的研究には、全ゲノムスクリーニング、候補遺伝子研究、染色体再編成研究、突然変異分析、および比較ゲノムハイブリダイゼーション研究が含まれている。総合すると、これらの研究は、シナプス形成と維持、形態形成、およびカルシウム調節とシグナル伝達に関与する遺伝子を含む、3つの主要なシステムに関与する遺伝子を含む自閉症感受性の新たな状況に貢献してきた。
シナプス形成と維持
おそらく、自閉症の感受性遺伝子の同定における最大のブレークスルーは、脆弱X症候群、結節性硬化症、およびレット症候群を含む、自閉症またはASDに関連する臨床的特徴を示すが、より単純な遺伝パターンを持つ疾患の研究から得られたものである。一般に、これらの疾患に関連する遺伝的欠陥は、シナプス形成および維持に影響を与える。自閉症症例の3%から4%を占める脆弱Xは、Xq27.3にあるFMR1遺伝子の5’領域における不安定なトリヌクレオチドリピートによって引き起こされる。このリピートは、次世代に伝達されるにつれて拡大し、FMR1の異常なメチル化および発現抑制をもたらす。FMR1は、核から細胞質へのRNA輸送のシャペロンとして機能し、シナプスでのmRNA翻訳に関与するRNA結合タンパク質を産生する。脆弱Xの個人およびこの疾患のマウスモデルにおいて、樹状突起スパイン密度(正常より増加)および形態(正常より長く細い)の異常が報告されている。おそらく自閉症症例の2%から10%を占める結節性硬化症(痙攣性疾患を持つ自閉症の個人では結節性硬化症の割合が高い)は、2つの腫瘍抑制遺伝子のいずれか、9q34上のTSC1、および16p13上のTSC2の変異に起因し、どちらもグアノシン三リン酸加水分解酵素(GTPase)不活化に関与している。マウスにおけるTSC1の単一コピーの喪失は、細胞骨格動態および樹状突起スパイン構造を破壊することが示されている。 ニューロリギン(NLGN)、SHANK、およびニューレキシン遺伝子ファミリーは、シナプス形成において役割を果たし、自閉症に罹患した一部の個人で観察される変異の影響を受ける可能性のある追加のリスク遺伝子座である。X染色体上に位置するNLGN遺伝子は、シナプス後グルタミン酸作動性ニューロンに位置する細胞接着分子を産生する。げっ歯類で変異すると、これらの遺伝子は輸送不良およびシナプス誘導の欠陥を示す。変異していない形態では、それらの発現は軸索における正常なシナプス前終末の形成を誘導する。変異体では、シナプス膜への小胞輸送の欠陥およびシナプス誘導の減少が見られる。NLGN3のヒト変異を持つマウスは、社会的新規性選好の障害および発声の減少を特徴とする興味深い表現型を示し、これらはヒトにおけるこの障害の中心的な特徴の2つである。SHANK遺伝子ファミリーは、シナプス後膜に位置し、PSDのマスターオーガナイザーとして機能する足場タンパク質をコードする。SHANK3は、SHANK遺伝子ファミリーの3つのメンバーの1つであり、フェラン・マクダーミド症候群として知られる22q13欠失症候群の最小領域に位置し、顕著な自閉症的特徴を含む。より大規模な研究では、SHANK3変異が全自閉症被験者の約0.85%に存在することが判明している。自閉症に関連するシナプス関連遺伝子の3番目のファミリーはニューレキシンである。ニューレキシンはニューロリギンと相互作用し、シナプス前ブートンの形成に不可欠である。ニューロキシンはまた、カルシウムチャネルをシナプス小胞に結合させ、エキソサイトーシスを介して神経伝達物質放出を媒介する。ニューロリギンおよびSHANK3と同様に、ニューレキシン(NRXN1)の変異はまれに発生するが、自閉症の個人の独立した家族系統で複数回出現している。
形態形成
症候性自閉症の多くの個人は、顔面異形および/または異常な身体成長率を示す。自閉症児の約20%は、人生の最初の数年間に頭部成長の軌跡が増加した大頭症を示すが、より少数の患者は小柄症を示す。ほとんどの場合、異常な成長パターンの病因はまだ明らかではないが、症候性自閉症のいくつかの形態は、胚発生および器官形成に関与する遺伝子のまれな変異と関連付けられている。脳幹、小脳、脳神経、および頭蓋冠の一部の正常な発達に必要な遺伝子であるHOXA1の変異が、自閉症的特徴を含む異種性の表現型を持ついくつかの家族で同定されている。染色体10q23上の腫瘍抑制遺伝子PTENにミスセンス変異が同定されている。PTEN変異は、カウデン症候群、バナヤン・ライリー・ルバルカバ症候群、プロテウス症候群、およびレルミット・デュクロ病を含む広範な症候群群と関連付けられている。PTENは、細胞増殖および身体成長を調節するために栄養利用可能性および成長因子シグナルに関する情報を取り込むために重要なERK/P13K/mTOR経路で相互作用することが示されている。PTENマウスノックアウトモデルは、大頭症、CNS過成長、および過剰な樹状突起および軸索成長を示す。PTEN変異を伴う症候群は、癌のような非精神医学的表現型を含む異種性の特徴を有し、特定の遺伝的変異の最終的な発現が遺伝的背景(他の変異)および環境要因の複雑な相互作用に依存することを強調している。 自閉症の別の症候性形態はレット症候群であり、これはX連鎖広汎性発達障害(既知の遺伝的病因を持つ最初のもの)であり、女児にのみ発生し、正常な早期発達に続いて、特に4歳までに社会的関与および目的のある手のスキルの喪失を伴う。この症候群はまた、ASDおよびASD様障害におけるシナプス形成および維持の異常も指摘している。レット症候群は、遺伝子発現およびクロマチン構造を調節し、正常な脳発達に必要なメチル化DNA結合タンパク質を作るMeCP2の変異によって引き起こされる。MeCP2の喪失は、神経細胞成熟の遅延および異常なシナプス形成と関連している。MeCP2の機能喪失変異は男性では致死的であり、女性の約70%で重篤な症状を引き起こすが、一部の変異は、ある程度の知的障害および/または言語障害を伴う比較的軽度の表現型をもたらす。症候性表現型に対するこれらの変異の異種性の発現は、症状の最終的な顕在化を決定する上で遺伝的背景が果たす重要な役割を再び強調している。
カルシウム調節とシグナル伝達
自閉症の少なくとも一部のケースと関連付けられている遺伝子群には、イオンチャネル、受容体、およびCa2+調節シグナル伝達分子が含まれます。構成的に活性なチャネルを発現し、過剰なCa2+流入を引き起こすいくつかのL型電位依存性Ca2+チャネルの機能獲得型変異は、自閉症と関連付けられています。Timothy症候群は、L型電位依存性Ca2+チャネルであるCACNA1Cの機能獲得型変異によって引き起こされ、知的障害と自閉症を特徴とします。興味深いことに、CACNA1Cは統合失調症や双極性障害と関連付けられており、診断カテゴリーを横断する遺伝的リスク要因の例を提供しています。別のL型電位依存性Ca2+チャネルであるCACNA1Fの機能獲得型変異は、自閉症的特徴を伴うこともある先天性夜盲症の原因です。カルシウム調節と関連付けられている他の遺伝子でも、いくつかの遺伝子変異が特定されています。ある自閉症のケースでは、カルモジュリンによって調節されるナトリウムチャネルを発現するSCN2Aに変異が特定されました。この変異は、カルモジュリンとチャネル間の結合親和性を阻害し、脱分極中の持続的なチャネル活性につながります。別のケースでは、脱分極または細胞内Ca2+によって活性化されるカリウムチャネルをコードするKCNMA1を破壊する染色体転座が特定され、構成的に脱分極した静止膜電位と異常な神経興奮性につながります。
自閉症における一般変異の役割
前述のセクションで述べた遺伝子変異はまれであり、自閉症のケースの合計で10%未満しか説明していません。遺伝子型が判明している大規模な個人サンプルでは、疾患の責任の40%から60%を一般変異が説明すると推定されており、2019年には、多くの力不足の遺伝子マッピング調査が有意な知見を特定できなかった後、国際コンソーシアムが約20,000のASDケースと30,000の対照のサンプルを収集し、最初の堅牢なGWA(ゲノムワイド関連解析)研究の知見を特定しました。それは、疾患の5つの再現されたリスクアレルでした。胎児および成人の脳からのトランスクリプトームデータの分析により、ASDリスク遺伝子座が、家族性ではない自閉症のケースに見られるまれな、および/またはde novo変異によって影響を受ける分子経路(PTBP2、CADPS、KMT2Eなど)と関連していることが示され、家族性および非家族性の疾患ケース間の共通の細胞生物学が実証されました。
上記の同じASD GWA研究は、ASDサンプルと、学業成績に関する表現型を含む大規模な独立したサンプルを統合し、ASDサンプルがより高い学業成績と知能に関連する遺伝的要因を共有していることを発見しました。知能とASD診断の間の共通の遺伝的影響の発見は、ASDの高機能個人(例:アスペルガー症候群と分類された個人)の間でのみ見られ、高機能自閉症スペクトラム障害がその遺伝的構造に基づいて他の広汎性発達障害と区別できることを示しています。実際、他の遺伝学的研究の結果と組み合わせると、自閉症および他の発達障害の新たな全体像は、多くの遺伝性の高い精神疾患が知能と正の相関があり、小さな影響の共通遺伝的要因からの広範な多型性の寄与があることを示しています。これらは、遺伝性が低く(すなわち、遺伝的対立遺伝子の影響が少ない)、認知機能により大きな影響を与えるde novo変異の有病率が高い、散発性広汎性発達障害の異質なクラスとは病因的に異なるものです。
まとめ
自閉症の遺伝学的調査は、ここ数年で著しく進歩しました。自閉症遺伝学の成功は、主に次の3つの要因に起因すると考えられます。(1)この疾患の多くの形態の高い遺伝性、(2)広範な調査のために自閉症家族の大規模サンプルを科学コミュニティに容易に利用できるようにした国際的な協力、および(3)連鎖研究、既知の染色体異常の調査、比較ゲノムハイブリダイゼーション、変異解析、および自閉症に関連する表現型を示す動物モデルの調査を含む補完的なアプローチを使用して得られた比較的整合性のある知見です。統合失調症、双極性障害、大うつ病などの他の精神疾患と比較して、ASDの病因に関与する分子生物学と細胞経路の比較的精緻なモデルが現在存在し、これらは疾患の家族性および非家族性バージョン間で有意な重複があるようです。この進歩は、大部分が大きな影響を与えるまれな変異によって引き起こされる症候性自閉症のケースの調査に起因しますが、新たな証拠は家族性ケースの遺伝的構造を明らかにし始めています。興味深いことに、十分に強力な家族性自閉症のGWA研究は、症候性ケースと家族性ケースの間には共通の細胞経路のセットがあることを示していますが、認知機能と知能に関連する遺伝的要因については、家族性ケースの遺伝的基盤が症候性ケースから分岐することも示しています。ASDの家族性ケースは、より高い知的機能に寄与する遺伝子と関連していますが、非家族性ケースは、より低い知的機能に素因となるde novo変異の高い割合と関連しています。最近の進歩は啓発的であり、さらに多くの発見があります。現在ASDと関連付けられている遺伝子座は、総遺伝性のほんの一部しか説明しておらず、発見されるべき多くの遺伝子座が残っていることを示しています。
双極性障害
双極性感情障害の遺伝的基盤を探る試みは、多くの誤解と部分的な回答に満ちていました。双極性障害の遺伝子マッピングの試みの歴史は、精神疾患の極端な複雑さだけでなく、これらの疾患に対する遺伝学的アプローチの進化も示しています。数十年にわたって実施された数多くの遺伝疫学的調査は、双極性障害のリスクに対する遺伝的寄与を強く支持してきました。双極性障害の一致率の現在の推定値は、一卵性双生児で65%から100%、二卵性双生児で10%から30%の範囲であり、この疾患が非常に遺伝性が高い(約60%から80%の間)ことを示しています。いくつかの研究は、双極性障害が単極性大うつ病よりも実質的に遺伝性が高いことを示しており、単極性大うつ病の推定遺伝率は30%から40%です。初期の家族研究は、双極性障害の分離パターンが、主要な効果を持つ遺伝子座の単一遺伝子遺伝と両立可能であることを示唆していました。このような遺伝子座が分離する双極性障害の家系が存在する可能性はありますが、そのような家系が存在するとしても、非常にまれであるという証拠が増えています。さらに、遺伝子連鎖研究が、いかなる家系においてもそのような遺伝子座を明確な証拠をもって発見できなかったという事実は、この可能性に反しています。一卵性同胞から第一度近親者への双極性障害の再発リスクの急速な減少も、単一遺伝子遺伝モデルとは一致せず、むしろ複数の相互作用する遺伝子のモデルを示唆しています。
1987年にX染色体と11番染色体における双極性障害への連鎖に関する最初の報告がなされた後、大きな興奮が続きました。研究者たちは、いくつかの家族で、双極性障害や他の感情障害がX連鎖様式で遺伝しているように見えることに注目しました。同様に、これらの障害は、色覚異常やG6PD欠損症(X染色体にマッピングされる)を持ついくつかのイスラエル人家族で共分離しているように見えました。色覚異常またはG6PD欠損症をマーカー遺伝子座としてこれらの家系で実施された連鎖研究は、4から9のLODスコアを示しました。11番染色体の初期の研究は、X染色体の研究と同様に、少数のマーカーを単一の領域でテストした後、有意な連鎖を報告しました。この場合、双極性障害に強く関係する大家系であるオールドオーダーアーミッシュの家系でした。当然のことながら、これらの発見は大きな関心を集めました。両方の研究は高いLODスコアを示し、連鎖の明確な証拠を提供しているように見えました。しかし、他の集団での再現研究は、X染色体と11番染色体のいずれについても陽性結果を生み出すことに失敗し、連鎖が元々報告されたサンプルで、家系に追加の罹患者を含めるように拡張された場合や、推定連鎖領域で追加のマーカーがタイピングされた場合、これらの両方の染色体領域における連鎖の証拠は実質的に消失しました。それぞれのケースで最も可能性の高い説明は、元の連鎖結果が偽陽性であり、後から見れば比較的乏しい証拠の過度に楽観的な解釈を反映していた可能性があるということです。
双極性障害の初期の連鎖研究は、利用可能なマーカーが少なかったため、ごく少数のマーカーしか評価しませんでした。1990年代にゲノムの遺伝子連鎖地図が構築されると、双極性障害を含むほとんどの複雑形質の連鎖研究は、ゲノムワイドに探索を開始しました。ゲノムワイドマッピング研究の利点は、特定の表現型の生物学的根拠に関する事前知識を必要としないことです。完全なゲノムスクリーニングは、偏りなくゲノム内のすべての点で連鎖の証拠を評価する機会を提供します。ゲノムワイド研究は、恣意的な場所の少数のマーカーや少数の候補遺伝子に焦点を当てた研究よりも、真の連鎖を検出する力が明らかに大きかったにもかかわらず、これらの調査も一般的に期待外れの結果に終わっています。双極性障害やその他の複雑形質に対する再現性のある有意な連鎖結果を達成することの難しさは、18番染色体上に双極性障害感受性遺伝子座を示唆しているものの、明確に証明されていない多くの遺伝子マッピング研究をレビューすると明らかです。
連鎖の最初の報告は、18番染色体の11個のマーカーを調べ、セントロメア付近で示唆的な連鎖を特定した部分ゲノムスクリーニングから来ました。双極性障害の遺伝パターンは不明であるため、結果は劣性モデルと優性モデルの両方を使用して分析されました。一部のマーカーは、一部の家族では劣性モデルで陽性であり、一部の家族では優性モデルで陽性であり、一部のマーカーは両方のモデルで家族のサブセットで陽性のLODスコアを示しました。この発見を他の集団で再現する試みはまちまちでした。これまでのところ、少なくとも2つのグループは、彼らのサンプルで18番染色体のセントロメア周囲領域への連鎖の証拠を見つけていませんが、別の1つのグループはこの領域への連鎖を支持する証拠を見つけています。他の研究では、2つの大規模なコスタリカ人家族における完全なゲノムスクリーニングを含め、18番染色体上で示唆的な連鎖の証拠が見つかっており、18q22-23の領域だけでなく、18pの領域でも連鎖の証拠が得られています。これらのいくつかの研究の複合的な証拠は、やや矛盾していて混乱していますが、18番染色体上に少なくとも2つの異なる感受性遺伝子座があることを示しています。1つは18p上、もう1つは18q上です。
上記で概説した18番染色体に関する複雑さは、遺伝子マッピング研究で遭遇する潜在的な困難を浮き彫りにしています。小規模な研究で得られた有意な知見の再現は、異なる家族のサンプルではしばしば失敗します。小規模な連鎖研究の検出力の低さを克服する一つのアプローチは、メタアナリシスを用いて、個々の研究の数値を組み合わせて関心のある平均効果を推定することです。これまでのところ最大のメタアナリシスには約1,000家族が含まれていましたが、この大規模なサンプルでも、明確な連鎖シグナルが特定されたかどうかは依然として議論の余地があります。これらのメタアナリシスで知見が得られなかったことについては、さまざまな説明が提示されていますが、その筆頭は、統計的に検出力が不足していた可能性です。もう一つの考慮事項は、複数の小さな効果の遺伝子が相互作用して疾患を引き起こしている可能性であり、その場合、これらの遺伝子は連鎖アプローチでは検出が困難になります。別の可能性としては、各家族が固有の遺伝的変異のセットを分離しており、連鎖解析ではこれらの状況でリスク遺伝子座を特定する検出力が低いという点です。最終的に、双極性障害の遺伝的構造は、一般集団で分離する一般的な変異と、最近の祖先に特有で比較的少数の家族内でのみ分離する稀な対立遺伝子の組み合わせを含むことが判明するかもしれません。精神医学的遺伝学における最近の遺伝子マッピングの努力の多くは、GWA(ゲノムワイド関連解析)手法を用いて、小さな効果の一般的なリスク遺伝子座を特定することに焦点を当ててきました。数十年にわたるフラストレーションのたまる連鎖研究と比較して、最近のGWA調査はかなりの成功を収めています(後述)。しかし、GWA研究で特定された個々の一般的な変異は、疾患リスクのほんの一部(約1%〜2%)しか説明していません。したがって、家族ベースの連鎖手法は、一般的なリスク変異を背景に発生する稀なリスク変異を特定し、どの個人が疾患を発症するかを決定することで、精神疾患の遺伝的構造の完全な解明において潜在的に重要な役割を担っています。
上記で述べた連鎖解析の期待外れな結果とは対照的に、双極性障害のGWA研究は、統計的に厳密な有意な知見をもたらし始めています。2007年、ウェルカムトラストケースコントロールコンソーシアム(WTCCC)は、2,000のケースと3,000のコントロールにおいて約500,000のSNPをタイピングした双極性障害の最初のGWA研究を実施しましたが、ゲノムワイドに有意なリスク変異は特定されませんでした。この陰性結果は、WTCCCに他の6つの一般的な疾患が含まれており、それらすべてが何らかの有意な知見を生み出したことを考えると注目に値しました。2008年、同様の規模の調査が、Systematic Treatment Enhancement Program for Bipolar Disorder and University College of London (STEP and UCL) のサンプルで実施されましたが、これもゲノムワイドに有意なリスク遺伝子座を特定できませんでした。その後、WTCCC、STEP、およびUCLはデータを統合して、4,387のケースと6,209のコントロールのサンプルサイズを得て、ついに10q21.2染色体上のANK3(アンキリンG)遺伝子内に存在するSNPについて、疾患に対する最初のゲノムワイドな有意な関連を報告しました。ANK3の知見はその後、追加の研究で複数回再現されており、2020年末までに約30の遺伝子座が双極性障害の確認されたリスク因子として確立されました。集団ベースのGWAの知見として予想されるように、リスク対立遺伝子の効果量は低い(オッズ比1.1〜1.3)ため、個々にこれらの一般的な変異は疾患リスクのわずかな増加にしか寄与しないことを示しています。
リスク遺伝子座によって影響を受ける遺伝子と生物学的経路の決定的なリストにはさらなる作業が必要ですが、いくつかの知見には確信が高まっています。ANK3遺伝子は複数のGWA研究で特定されており、脳組織に高度に発現し、ニューロン軸索の構造に関与するアンキリンBをコードしています。双極性障害と関連するSNPのいくつかは、アンキリンBの発現レベルとも関連しています。マウスの前脳の錐体ニューロンにおける遺伝子の条件付きノックアウトは、軸索の電位依存性ナトリウムチャネルおよびカリウムチャネルの構造に破壊をもたらします。興味深いことに、ノックアウトマウスは躁病に似た行動障害を示し、これはリチウムで修正されました。双極性障害と強く関連する2番目の遺伝子はCACNA1C遺伝子で、これは自閉症(上記参照)、うつ病、統合失調症(下記参照)とも関連しています。CACNA1Cは、神経発達とシナプス機能に関与するL型電位依存性チャネルをコードしています。双極性障害と強く関連する3番目の遺伝子はTRANK1で、これも統合失調症と関連しており、血液脳関門の生理機能に関与している可能性があります。双極性障害と関連するほとんどのリスク遺伝子座は、生物学的機能と確固たる関連がありません。パスウェイ解析は、遺伝子オントロジーデータベースにおける増え続ける証拠を活用して、遺伝子を特定の生物学的経路にグループ化し、イオンチャネル病、内分泌調節、転写後修飾など、双極性障害におけるさまざまな細胞プロセスの関与の証拠を提供しています。わずか5年前には不明瞭だった双極性障害の遺伝的構造の現在の理解は、明確になり始めており、家族ベースの研究と中間表現型アプローチと組み合わせて、明確なメカニズム的疾患モデルにつながることが期待されます。
統合失調症
双極性障害と同様に、統合失調症に関連する遺伝子変異のマッピングは、強い遺伝疫学的証拠による遺伝性の確認から始まり、その後、曖昧な結果を生み出した家系ベースの連鎖研究が続き、そしてGWA(ゲノムワイド関連解析)研究を通じて、一般的で小さな効果の変異が急速に発見される期間へと続きました。2021年現在、200を超える再現性のあるリスク変異があり、国際コンソーシアムを通じてサンプルサイズを増やし続ける計画があります。遺伝子マッピングが成功する以前、家族研究では、統合失調症が遺伝的要因に強く影響され、遺伝率が60%から80%の間であると明確に確立されていました。1990年代初頭、家族ベースの連鎖研究は、この疾患に関連する染色体領域の特定を開始しました。6p24-22染色体は、統合失調症の全ゲノムスクリーニングによって最初に示唆された領域の一つであり、この場合は統合失調症に強く関係するアイルランドの家族の研究から得られました。さらに6つの連鎖研究がほぼ同じ領域で陽性結果を示しましたが、少なくとも3つの研究はその領域との連鎖を見出していません。元のアイリッシュの親族における関連解析を用いたこの領域の精密マッピングは、ジスビンディン(DTNB1)を統合失調症の候補遺伝子として提案することにつながりました。ジスビンディンの追加の関連研究は曖昧でした。個別の連鎖研究はその後、候補遺伝子であるDISC1およびDISC2(統合失調症で破壊された1および2)を含む1番染色体上の領域を指し示しました。これらは1q21-22および1q32-42染色体上に位置します。これらの遺伝子は、1990年代初頭に大規模なスコットランドの家系で最初に特定されました。この家系では、1番染色体と11番染色体の間の均衡型転座が分離しており、重篤な精神疾患と関連している可能性がありました。DISC1および2は、染色体転座の切断点近くに位置していたため、元のスコットランドの家族で特定されました。ジスビンディンと同様に、DISC1および2の追跡調査は曖昧でした。
上記の連鎖研究とほぼ同時期に、大規模なアイスランドの親族に焦点を当てたゲノムワイドスクリーニングにより、8p21-22染色体上に統合失調症の候補領域が特定されました。この領域の精密マッピングにより探索が絞り込まれ、最終的にニューレグリン1(NRG1)が統合失調症の候補遺伝子として提案されました。関連研究は再び曖昧で解釈困難な結果をもたらしました。元の研究で関連を示したSNPマーカーを用いた14の個別の研究のメタアナリシスは、追跡調査研究間で有意な異質性を示しました。また、マーカーSNPによって「タグ付けされた」特定のリスクアレルと異なる集団における統合失調症との間に一貫した関連がないことも示されました。しかし、各関連研究の統計的検出力を考慮に入れた後、メタアナリシスは、遺伝子レベルでのNRG1と統合失調症の間に正の関連を示しました(SNPまたはハプロタイプレベルとは対照的に)。
曖昧な遺伝学的研究にもかかわらず、ジスビンディン、DISC1、およびニューレグリンの機能的産物の分子および神経生理学的調査にかなりの資源が投入されてきました。これら3つの遺伝子のそれぞれについて変異マウスが利用可能になり、興味深い生物学的知見を示すために使用されています。例えば、ジスビンディンは海馬と背外側前頭前野に発現しています。ジスビンディンタンパク質はβ-ジストロブレビンと結合し、シナプス構造とシグナル伝達に関与しているとされています。DISC1は細胞研究で神経突起形成に影響を与えることが示されており、DISC1の変異マウスは、学習、記憶、社会性を含む幅広いテストで障害を示します。ニューレグリンは、シナプス形成、ニューロン遊走、神経伝達など、多数の機能を仲介する成長因子ファミリーに属しています。ニューレグリンのシナプス後標的であるerbB4の標的破壊は、シナプスグルタミン酸作動性機能低下につながります。発見された興味深い生物学にもかかわらず、これらの遺伝子のいずれかがヒトの統合失調症の病因にどの程度寄与しているかは不明のままであり、多くの遺伝学者は、現在の候補遺伝子のリストから生成された変異マウスを精神疾患のモデルとして正当性を慎重に支持しています。
他の精神疾患と同様に、統合失調症のGWA研究は、2009年まで期待外れな結果をもたらしました。この年、国際統合失調症コンソーシアム、統合失調症の分子遺伝学、および精神医学ゲノミクスコンソーシアムの統合失調症ワーキンググループがサンプルを統合し、6p22染色体上の主要組織適合性複合体(MHC)のSNPを疾患のリスク遺伝子座として特定しました。統合失調症のリスク遺伝子座をMHC領域にマッピングすることは画期的な発見でしたが、この知見の完全な意義を解釈することは困難です。母体感染への曝露が統合失調症のリスク因子であることは以前から知られていました。したがって、特定のMHCの構成が、脳の発達および/または疾患に素因となるプロセスに影響を与える異常な免疫応答につながる可能性があります。さらに、ニューロンはMHC分子を発現しており、そこでシナプス形成、神経突起伸長、長期可塑性など、脳の発達のいくつかの側面に関与しています。したがって、MHCの遺伝的変異は、免疫調節メカニズム以外で脳の発達に直接影響を与える可能性があります。
MHCがリスク遺伝子座として発見された直後、GWA研究は1ダース以上の追加のリスク遺伝子座を特定し、サンプルサイズが増加するにつれて、新規および再現されたリスク遺伝子座の数が増加しました。2014年、精神医学ゲノミクスコンソーシアムの統合失調症ワーキンググループは、当時37,000のケースと113,000のコントロールのサンプルサイズに達していましたが、統合失調症のリスクに寄与する108の異なる遺伝子座を特定しました。
特定のSNPで遺伝的関連性が特定されたとしても、その遺伝子型を持つSNPが因果的な対立遺伝子であるとは限らないことに注意することが重要です。実際、現在のほとんどの遺伝子型判定プラットフォームでタイピングされたSNPが機能的な意義を持つ可能性は低いでしょう。タイピングされたSNPは、因果的な変異体のごく近傍(約100kb)で連鎖不平衡(LD)にある可能性が高いです。さらに、比較的小さな領域内でも広範な配列多様性があることを考えると、特定の変異体または変異体のセットを複雑な疾患の発症における因果的因子として確立することは容易ではありません。変異体の明確な特定を複雑にしているのは、GWA研究によって特定されるリスク遺伝子座の多くが、メンデル遺伝子の大効果と比較して、遺伝子機能のより微妙な側面に影響を与える因果的変異体を保有すると予想されることです。メンデル変異がしばしば遺伝子のコーディング配列に大きな破壊をもたらすのに対し、小さな効果の一般的なリスク変異は、発現または調節のレベルで遺伝子機能に影響を与えると考えられています。タンパク質コーディング領域に影響を与える変異とは対照的に、遺伝子のイントロン/エキソン構造の外側にあって、発現やクロマチン構造を制御する調節領域に影響を与えている可能性のある変異を特定することはより困難です。ENCODEプロジェクトのような進行中のイニシアティブは、調節タンパク質結合部位やオープンクロマチン領域を含むゲノムの生化学的に活性な領域を特徴づけることを目的としており、それによって非コーディング領域における変異の解釈を導くための貴重なリソースを提供しています。
統合失調症の病因に関与する特定の生物学的経路の解明には、因果変異の明確な特定と、それらが特定の遺伝子に与える影響が必要ですが、最近のリスク遺伝子座の蓄積は示唆的な方向性を提供しています。リスク遺伝子座は、すべての細胞プロセスに関与する遺伝子間にランダムに分布しているようには見えず、むしろ、脳機能にとって重要なより小さなサブセットのプロセスに関与する遺伝子付近に濃縮されているように見えます。これには、神経発達(3q26.33のFXR1、2q33.1のSATB2、7q32-q33のPODXL、14q32.2のBCL11B、9q21.32のTLE1、15q22のTLE3、1q24のBRINP2)、グルタミン酸作動性神経伝達(16p13.2のGRIN2A、5q33.2のGRIA1、17p13.3のSRR、4q33のCLCN3、7q21.12のGRM3)、ニューロンカルシウムシグナル伝達(12p13.3のCACNA1C、10p12のCACNB2、12q24.2のCAMKK2、11q24のNRGN、12q24.11のATP2A2、22q13.1のCACNA1I、6q12-13のRIMS1)、およびシナプス機能と可塑性(16p11.2のKCTD13、Xp21.33-32のNLGN4X、11q25のIGSF9B、3p26.3のCNTN4、5q14.3のMEF2C、7q33のPTN、Xp22.12のCNKSR2、15q14のPAK6、6q14.2のSNAP91)が含まれます。
過去数年間でGWA研究は大きな成功を収めましたが、一般集団で分離する一般的な変異だけでは、精神疾患への推定される遺伝的寄与のすべてを説明できないことが明らかになりました。この事実は「失われた遺伝率」問題として知られています。例えば、双生児研究や家族研究に基づくと、統合失調症の遺伝率は約70%ですが、一般的な遺伝的多型によって説明される総分散は約30%と推定されており、説明されていない遺伝率の「ギャップ」が存在します。失われた遺伝率問題の原因については、さまざまな説明が提示されています。失われた遺伝率の少なくとも一部を説明する可能性が高い要因の一つは、GWA手法では検出できない中〜大効果の稀な遺伝子が罹患した個人に存在している可能性です。他のすべての精神疾患と同様に、統合失調症の遺伝子マッピングの将来の方向性には、(1)集団GWA研究の検出力を高めるためにこれまでよりも大規模なサンプルセットを蓄積する継続的な努力、(2)単一の個人に存在する(すなわち、de novo変異)または拡張家族内で分離する中/大効果の稀な変異を特定すること、および(3)中間表現型アプローチを用いて疾患の病因に関与する生物学的経路の遺伝的構造を特徴づけることが含まれるでしょう。
大うつ病
大うつ病は最も一般的な精神疾患であり、世界的に疾病負荷の主要な原因の一つです。生涯有病率は国によって異なりますが、一般的に**8%から12%**の範囲です。大うつ病への遺伝的寄与の証拠は、発端者の第一度近親者における疾患のリスクが一般集団の2〜3倍高いことを示す家族研究から来ています。大うつ病の遺伝率は、他の気分障害や精神病性障害よりも一般的に低く、30%から40%と推定されています。大規模な集団研究は、女性における疾患の有病率が高いことを一貫して示しており、双生児研究は、この疾患が実際に女性においてより遺伝的影響を受けていることを示しています。女性の推定遺伝率は約40%であるのに対し、男性は約30%です。これらの同じ研究は、大うつ病の遺伝的原因の大部分が男性と女性の間で共有されているものの(性別間の遺伝的相関は0.6〜0.9と推定される)、遺伝的要因のかなりの部分が性特異的であると推定しています。性別間の遺伝的差異に加えて、うつ病のサブタイプ間にも遺伝的差異があるようです。例えば、反復性うつ病は単一エピソードのうつ病と比較してより高い遺伝的影響(遺伝率40%対30%)を持っているようであり、産後うつ病は非産後うつ病と比較してより高い遺伝性(50%対30%)を持っています。性別および疾患サブタイプ間の遺伝的影響の異なる推定値は、疾患の異質性と、多くの複雑な相互作用する生物学的および環境的要因が、各個人における疾患の発現または回復力に影響を与えるという事実を浮き彫りにしています。
GWA(ゲノムワイド関連解析)研究が実施される以前、遺伝学研究は主に、うつ病に影響を与える候補遺伝子のテストに焦点を当てていました。40年前から、研究者たちは、うつ病の根底にある生物学に関連すると仮説が立てられた、神経伝達物質および関連する代謝経路に関連する特定の遺伝子または少数の遺伝子群のテストを開始しました。過去数十年にわたり、200を超える候補遺伝子を対象とした1,500以上の調査が発表されましたが、矛盾した、混乱させるような結果に終わっています。最近のGWA研究は、候補遺伝子研究で報告された有意な関連性を特定するのに十分な検出力を持っていますが、一貫してそうすることに失敗しています。非常に十分な検出力を持つデータセットで、最も一般的に研究されている候補遺伝子のうち18個に焦点を当てた分析では、DRD2(ドーパミン受容体D2)遺伝子の証拠が見つかりましたが、他のどの遺伝子の証拠も発見できず、これまでに報告された「有意な」候補遺伝子の知見の圧倒的多数が偽陽性であることを示しています。
他の神経精神疾患と同様に、大うつ病の遺伝的構造には、中〜大効果の稀または新規の変異と、多数の小さな効果の一般的な遺伝子座が混在すると予想されます。これまでのところ、家族ベースの連鎖研究では、疾患に寄与する稀な変異について強力な知見は特定されていません。最近の全エクソームおよび全ゲノムシーケンシング研究は、大うつ病の個人が有害な単一エクソン変異の負担が高いことを示唆する証拠を提供していますが、これらの知見を確認するにはより大規模なサンプルサイズが必要となるでしょう。また、エンハンサー領域にマッピングされた**短い欠失CNV(コピー数変異)**が大うつ病の個人で豊富であるという示唆的な証拠もあります。CNVとエクソン変異に関する示唆的な証拠にもかかわらず、新規および稀な変異の特性評価には、より大規模なデータセットとより洗練された分析方法(例:疾患サブタイプおよび中間表現型の分析)が必要となるでしょう。
稀な変異の探索の失敗とは対照的に、大うつ病のGWA研究は実を結び始めています。しかし、統合失調症のような他の精神疾患と比較すると、大うつ病の低い遺伝率と高い異質性により、はるかに大規模な発見サンプルが必要となっています。例えば、統合失調症のGWA研究は、約15,000のサンプルサイズで厳密な成功を収め始め、平均して約250の新しいケースが追加されるたびに新しい遺伝子座を特定しています。対照的に、大うつ病の最初の厳密な遺伝学的知見には75,000のケースが必要であり、新しいリスク遺伝子座の発見には平均して約1,500の新しいケースが必要です。最初の成功したGWAは、2016年に直販型企業23andMeによって、自己申告のうつ病診断を用いて達成されました。その後数年間で、精神医学ゲノミクスコンソーシアムおよびミリオンベテランプログラムによるGWA研究が確認されたリスク遺伝子座のリストに追加され、現在、100を超えるリスク遺伝子座が特定されています。全体として、最近のGWA研究によって説明される総分散は、疾患に対する推定される遺伝的寄与全体のわずか約3%しか説明しておらず、大うつ病の高度な多遺伝子性と、サンプルサイズの増加および疾患サブタイプと中間表現型のより洗練された分析の必要性を再び浮き彫りにしています。
遺伝子レベルおよびパスウェイ解析は、ゲノム全体にわたる関連性を集約し、疾患に寄与する生物学的経路を探索するために使用されてきました。これらの分析は、シナプス機構、電位依存性カルシウムチャネル、グルタミン酸受容体、神経成長と分化に関与する遺伝子、免疫と炎症に関与する遺伝子を示唆しています。最も堅固に支持されている個々の遺伝子は、NEGR1(神経成長調節因子1)とOLFM4(オルファクトメディン4)です。これらの遺伝子座はそれぞれ、推定オッズ比1.04の小さな効果量を持っています。NEGR1は、神経突起伸長とシナプス形成に関与する細胞接着分子です。OLFM4は、炎症と免疫に関与する分泌型糖タンパク質です。確認された遺伝子座のセット内の他の遺伝子もこれらの生物学的プロセスに関与しており、小さな効果の一般的な変異が、シナプス機能、神経受容体、免疫/炎症を含む生物学的経路のサブセット内に蓄積し、個人の環境リスク要因への曝露に応じて、集合的に個人を大うつ病へと傾けるという新たな全体像を裏付けています。
不安障害
本来正常な感情である恐怖と不安の病的な形態は、最も一般的な精神状態の一つであり、生涯有病率は20%を超えます。DSM-V以前は、OCD(強迫性障害)と心的外傷後ストレス障害は不安障害のカテゴリーに含まれていましたが、それらの明確な特徴と生物学的メカニズムのためにカテゴリーから除外されました。不安障害には現在、全般性不安障害、パニック障害、恐怖症の診断が含まれます。不安障害の遺伝子マッピング研究は、他の精神疾患と同じパターンをたどっています。まず、疫学的調査が家族性集積の証拠を示し、次に、大部分が再現されていない遺伝子座を特定した家系ベースの連鎖研究と、矛盾した知見を生み出した多数の候補遺伝子研究が続き、そして最後に、大規模な集団サンプルで共通の変異の厳密な知見を生み出し始めたGWA研究が続きました。
家族における不安障害の疫学調査は、家族性集積に関する一貫した証拠を提供しており、患者の第一度近親者は不安障害のリスクが4〜6倍増加しています。双生児研究は、さまざまな不安障害の遺伝率が30%から50%の間であると推定しています。家族内のリスクには、広範な不安障害の発症が含まれており、異なる診断間でかなりの遺伝的重複があることを示唆しています。統計的に、モデリング手法は、比較的均一な共通の遺伝的要因が不安障害のコアセットに寄与しており、環境要因が疾患の最終的な形態に影響を与えることを確認しています。
不安障害の家系ベースの連鎖研究は、主にパニック障害の家族に焦点を当ててきましたが、最も強力なマッピングシグナルは、パニック、恐怖症、全般性不安症状、さらには頭痛のような医学的疾患を含む広範な表現型に対して得られています。1990年代後半から2000年代初頭にかけて、一連の調査により、1q、2q、7p、9q、13q、15q、22q染色体上で連鎖シグナルが特定されました。アルツハイマー病や双極性障害に関する同様の研究と比較して、家族における不安障害遺伝子のマッピングの取り組みは著しく少なく、これまでのところ、これらの知見のどれも再現されていません。連鎖研究が実施されていたのとほぼ同時期に、多くのグループが仮説に基づいた生物学的メカニズムに基づく候補遺伝子関連研究に焦点を当てていました。他の精神疾患の候補遺伝子研究と同様に、不安表現型の候補遺伝子研究は矛盾した結果を生み出し、この分野では現在、最近のGWA研究が大規模な集団サンプルを蓄積できるようになるまで、統計的に有意な知見を特定するにはサンプルサイズが非常に不足していたことを認識しています。
他の精神疾患のGWA研究と同様に、不安障害への最初の進出は検出力の低いサンプルサイズであり、再現性のある知見は生み出されませんでした。2016年、7つの先行研究のメタアナリシスは18,000を超えるケースのサンプルサイズを組み立て、3番染色体上のCAMKMT遺伝子内のSNPを含む2つのゲノムワイドに有意な遺伝子座を特定しました。その後まもなく、いくつかのグループが20,000を超えるサンプルサイズを組み立て、以前に神経症傾向と関連していた9番染色体上の領域を含む、さらに半ダースのリスク遺伝子座を特定しました。最も強力な生物学的支持を持つリスク遺伝子の一つは、信号伝達に関与するホスホジエステラーゼファミリー遺伝子であるPDE4Bです。このタンパク質の活性変化は、双極性障害や統合失調症で特徴づけられており、このタンパク質が欠損したマウスモデルは不安様行動を示します。これまでのGWA研究で最大規模のものは、ミリオンベテランプログラムで実施され、約20万人の個人のサンプルサイズでした。研究者たちは、遺伝子マッピングの表現型として、全般性不安障害2項目尺度(GAD-2)のスクリーナー質問を使用しました。GAD-2は特定の診断を提供するものではなく、不安感情の頻度と心配を止められない能力を問う2つの質問に基づいて、リッカート尺度で自己申告により不安の連続的な尺度を割り当てます。この表現型を用いて、この研究は5つの有意なゲノムワイド遺伝子座を特定しました。これには、遺伝子発現のグローバルレギュレーターであるSATB1遺伝子の近くの染色体上の1つと、エストロゲン受容体をコードするエストロゲン受容体遺伝子ESR1の近くの6番染色体上の1つが含まれます。MAD1L1遺伝子の近くの染色体上の3番目の遺伝子座は、以前に統合失調症と双極性障害のGWA研究で特定されていました。不安リスク遺伝子座と他の精神疾患との間の重複する知見は、疾患間の遺伝的重複を浮き彫りにしており、次のセクションの焦点となります。
精神疾患間の遺伝的重複
これまでのセクションでは、カテゴリーに定義された精神疾患がかなりの遺伝的異質性を示すことを強調してきました。これは、診断カテゴリーを特定する症状群が、異なる遺伝子セットの変異から生じる可能性があることを示しています。逆もまた真です。特定の遺伝子の変異は診断境界を越えることができます。初期の家族研究は、特定の疾患(すなわち統合失調症)と診断された個人の親族が、統合失調症だけでなく、大うつ病や双極性障害を発症するリスクも高いことを示しました。遺伝子マッピング研究は、遺伝的リスク遺伝子座が異なる疾患間で共有されていることを確認しています。例えば、カルシウムチャネル遺伝子CACNA1Cの変異は、自閉症、双極性障害、統合失失調症と関連付けられています。
大規模な個体サンプルにおける遺伝子型が利用可能になったことで、精神疾患と非診断的特性間の遺伝的相関を推定できるようになりました。2つの特性間の遺伝的相関(rg)は、家族データまたは集団ベースの遺伝子型から決定でき、多面的発現としても知られる、両方の特性に影響を与える共通の遺伝的要因の推定値を提供します。精神医学ゲノミクスコンソーシアムは、自閉症、注意欠陥多動性障害(ADHD)、双極性障害、大うつ病、統合失調症という5つのカテゴリーに定義された精神疾患の患者を含む大規模なサンプル(約30,000の症例と約30,000の対照)で、ゲノムワイドな共通変異の重複を調査しました。この調査により、統合失調症と双極性障害の間に顕著な重複が見られました。共通変異に起因する多遺伝子リスクの約68%が、これら2つの疾患間で共有されていました。大うつ病も他の疾患とかなりの重複を示し、共通変異に起因する遺伝的リスクの約45%が統合失調症と双極性障害の両方と共有され、約30%がADHDと共有されていました。統合失調症は、ASD(自閉症スペクトラム障害)と低い(約15%)ながらも有意な重複を示しました。自閉症は、双極性障害、大うつ病、またはADHDとは有意な遺伝的重複を示しませんでした。同様に、大うつ病との遺伝的重複を除いて、ADHDは他のどの疾患とも有意な重複を示しませんでした。これらの知見は、例えば統合失調症と双極性障害は区別が難しい場合があるが、双極性障害と自閉症は通常容易に区別できるという臨床シナリオと並行しているようです。これらの知見はまた、精神疾患の根底にある細胞経路の新たなパターンを示唆しています。一部の分子経路(例:カルシウムシグナル伝達)の変化は、神経システムを非特異的に乱し、最終的な精神症状が他の遺伝的および/または環境的要因によって決定される可能性があります。このシナリオは、共通の遺伝的リスク要因のかなりの割合を共有する統合失調症、大うつ病、双極性障害に特に当てはまるかもしれません。対照的に、ADHDのような疾患は、他の疾患ほど多くの共通の遺伝的リスク要因を共有せず、精神病性および気分スペクトラム障害につながるものとは異なる細胞経路の変化を通じて生じる可能性があります。
遺伝的相関は、精神疾患と非精神医学的特性間の遺伝的関係を理解するのにも役立ちます。例えば、不安のセクションで説明したGAD-2スコアのGWA研究は、うつ病(rg = 0.81)およびパーソナリティ特性である神経症傾向(rg = 0.75)と高い相関関係があります。同様に、大うつ病は神経症傾向(rg = 0.70)および主観的幸福感(rg = -0.75)と有意な遺伝的要因を共有しています。主観的幸福感との相関の負の推定値は、うつ病のリスク増加をもたらす遺伝的要因が幸福感に逆方向に影響を与えることを示しています。つまり、うつ病のリスクを増加させる遺伝子座は、主観的幸福感の尺度を低下させる傾向があります。大うつ病はまた、学業成績や認知能力とも負の遺伝的相関がありますが、パーソナリティ特性との相関よりははるかに低く、約rg = -0.15です。非精神医学的特性との遺伝的相関は、疾患のサブタイプを分析するのに役立ちます。例えば、非定型うつ病と肥満度指数(BMI)の間には、体重増加を経験する患者には共有される遺伝的要因があるように見えますが、食欲や体重の変化を経験しない患者には見られません。精神疾患と広範な医療および非医療特性間の遺伝的相関が利用可能になるにつれて、精神衛生に存在する複雑な異質性についてより明確な理解が深まるでしょう。これは、うつ病のような疾患にとって特に重要かもしれません。うつ病は、比較的一般的な症状群として現れる、多くの多様で独立したプロセスの共通の終点を反映しているからです。
将来の方向性
精神医学遺伝学の分野は移行期にあります。新しい技術は、複雑な行動の遺伝学的探求を現実のものにしました。35年前には、ヒト遺伝子マッピング研究は少数の個人における数少ないマーカーの使用を伴っていましたが、現在では数万人のサンプルで数百万のマーカーを遺伝子型決定することが日常的に行われています。高解像度神経画像診断のようなヒト神経系の表現型を評価するためのエキサイティングな新技術の開発、および精神医学研究における遺伝子マッピング研究でのこれらのアッセイの多数の実施は、精神医学研究における表現型と遺伝子型の大規模な関連付けのための並外れた機会を生み出しました。このような膨大で複雑なデータセットの分析は依然として大きな課題ですが、この目的のための方法論の開発は、現在、統計遺伝学およびバイオインフォマティクス研究の中心的な焦点となっています。
精神医学遺伝学の分野が進歩するにつれて、浮上してきたいくつかの主要な質問は、少なくとも部分的には、大規模なデータセットの分析を通じて間もなく回答されるかもしれません。1つの主要な質問は、精神疾患の遺伝的リスクが主に大きな効果の少数の稀な変異から派生するのか、それとも小さな効果の多数の一般的な変異から派生するのか(または稀な変異と一般的な変異のいくつかの組み合わせなのか)ということです。十分な検出力を持つGWA研究は一般的な変異を特定するはずですが、稀な変異を特定する検出力は非常に低いです。これまでのところ、連鎖研究が精神疾患の稀な原因変異を特定できなかったことは、やや効果の少ない稀な変異がこれらの疾患への感受性に重要な役割を果たす可能性を排除するものではありません。大規模なサンプルにおける全ゲノムシーケンシングは現在実行可能であり、精神疾患の遺伝的負荷が高い家族で分離する稀な変異を特定することを目標に実施されています。
2番目の質問は、精神病理学が現在のDSMの病理分類学のように離散的な疾患に分けられるのか、それとも症候群間の異質性と重複があまりにも大きく、現在の分類システムを徹底的に見直す必要があるのかという点です。一般的な遺伝的変異の集団ベースの調査からの推定値は、気分障害と精神病性障害が遺伝的リスク要因のかなりの重複を共有しているのに対し、自閉症やADHDのような疾患は、他の精神疾患とは遺伝的にさらに異なる可能性があることを示しています。この問題をより完全に詳しく説明するには、遺伝的リスク要因が他のリスク遺伝子や環境とどのように相互作用して特定の個人における症状を予測するかを調べ始める必要があります。さらに、主要な精神疾患の脳、行動、およびその他のエンドフェノタイプの遺伝学的調査にこの分野の焦点が増していることは、そのような革命の方向への一歩かもしれません。
3番目の主要な質問は、疾患の病因における遺伝子と環境の相互作用の役割を特徴づけることです。遺伝子-環境相互作用は、個々の遺伝的背景と環境の特定の特性との相互作用によって、特定の表現型が出現したり影響を受けたりする場合に発生します。遺伝子-環境相互作用を特定するには、遺伝子と表現型間の一次相関を特定するよりもさらに統計的検出力が必要であり、これまでに多くのそのような研究が発表されていますが、確固たる結論を出すには検出力が不足していました。しかし、人口統計学的および歴史的データの適切な評価を伴うサンプルサイズが増え続けるにつれて、遺伝子-環境相互作用の厳密な特定が可能になるでしょう。例えば、英国バイオバンクの50万人以上の個人を対象とした最近の調査では、タウンゼント剥奪指数によって測定された遺伝的および社会経済的データを用いて、(1)社会経済的スコアが不安、双極性障害、うつ病、自傷行為、および全体的な健康と関連していること、そして(2)社会経済的スコアに応じて疾患表現型に寄与する特定の遺伝子座と、遺伝子と環境の分析のゲノムワイドスキャンから示唆的な関連性が示されました。この解釈は、特定の遺伝子座が環境要因が存在する場合にのみ疾患リスクに寄与するというものです。社会経済学、トラウマ、物質への曝露、および精神衛生へのその他の既知または疑われる影響に関連するデータが体系的に蓄積されるにつれて、遺伝子-環境分析は、リスクと回復力に寄与する複雑な経路を特徴づけるための強力なツールを提供し、そのような疾患の病因と治療の両方を理解するための指針となるでしょう。
最後の質問は、予想される精神医学遺伝学的発見が健康アウトカム(予防または治療の改善のいずれかの観点から)に与える影響に関するものです。アルツハイマー病に関しては、具体的な進歩がなされており、この質問への回答は間もなく得られるかもしれません。この章で議論されている他の疾患については、過去数年間で急速な進歩が見られましたが、その新たな全体像は依然として流動的であり、回答可能な形でこの質問を組み立てるのに十分な理解が得られるまでには、まだ数年かかるかもしれません。
参考文献
- Balding DJ. A tutorial on statistical methods for population association studies. Nat Rev Genet. 2006;7(10):781-791.
- Bienvenu OJ, Davydow DS, Kendler KS. Psychiatric ‘diseases’ versus behavioral disorders and degree of genetic influence. Psychol Med. 2011;41(1):33-40.
- Cardno AG, Rijsdijk FV, West RM, et al. A twin study of schizoaffective-mania, schizoaffect-depresion, and other psychotic syndromes. Am J Med Gen B Neuropsychiatr Genet. 2012;159B(2):172-182.
- Chanock SJ, Manolio T, Boehnke M, et al; NCI-NHGRI Working Group on Replication in Association Studies. Replicating genotype-phenotype associations. Nature. 2007;447(7145):655-660.
- Civelek M, Lusis AJ. Systems genetics approaches to understand complex traits; Nat Rev Genet. 2014;15(1):34-48.
- Craddock N, Sklar P. Genetics of bipolar disorder. Lancet. 2013;381(9878):1654-1662.
- Flint J, Kendler KS. The genetics of major depression. Neuron. 2014;81(3):484-503.
- Flint J, Munafò M. Schizophrenia: genesis of a complex disease. Nature. 2014;511(7510):412-413.
- Dennison CA, Legge SE, Pardiñas AF, Walters JTR. Genome-wide association studies in schizophrenia: recent advances, challenges and future perspective. Schizophr Res. 2020;217:4-12.
- Glahn DC, Knowles EEM, McKay DR, et al. Arguments for the sake of endophenotypes: examining common misconceptions about the use of endophenotypes in psychiatric genetics. Am J Med Genet B Neuropsychiatr Genet. 2014;165B(2):122-130.
- Grove J, Ripke S, Als TD, et al; Autism Spectrum Disorder Working Group of the Psychiatric Genomics Consortium, BUPGEN, Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium, 23andMe Research Team. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51(3):431-44.
- Kellis M, Wold B, Snyder MP, et al. Defining functional DNA elements in the human genome. Proc Natl Acad Sci U S A. 2014;111(17):6131-6138.
- Kendall KM, Van Assche E, Andlauer TFM, et al. The genetic basis of major depression. Psychol Med. 2021;51(13):2217-2230.
- Lee SH, Ripke S, Neale BM, et al; Cross-Disorder Group of the Psychiatric Genomics Consortium, International Inflammatory Bowel Disease Genetics Consortium (IIBDGC). Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45(9):984-994.
- Meier SM, Deckert J. Genetics of anxiety disorders. Curr Psychiatry Rep. 2019;21:16.
- Munafò MR, Thiselton DL, Clark TG, Flint J. Association of the NRG1 gene and schizophrenia: a meta-analysis. Mol Psychiatry. 2006;11(6):539-546.
- Mutsuddi M, Morris DW, Waggoner SG, Daly MJ, Scolnick EM, Sklar P. Analysis of high-resolution HapMap of DTNBP1 (Dysbindin) suggests no consistency between reported common variant associations and schizophrenia. Am J Hum Genet. 2006;79(5):903-909.
- Ott J, Wang J, Leal SM. Genetic linkage analysis in the age of whole-genome sequencing. Nat Rev Genet. 2015;16:275-284.
- O’Tuathaigh CMP, Babovic D, O’Meara G, Clifford JJ, Croke DT, Waddington JL. Susceptibility genes for schizophrenia: characterisation of mutant mouse models at the level of phenotypic behaviour. Neurosci Biobehav Rev. 2007;31(1):60-78.
- Pinto D, Delaby E, Merico D, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94(5):677-694.
- Reiman EM, Webster JA, Myers AJ, et al. GAB2 alleles modify Alzheimer’s risk in APOE epsilon4 carriers. Neuron. 2007;54(5):713-720.
- Roybal K, Theobold D, Graham A, et al. Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci U S A. 2007;104(15):6406-6411.
- Rzhetsky A, Wajngurt D, Park N, Zheng T. Probing genetic overlap among complex human phenotypes. Proc Natl Acad Sci U S A. 2007;104(28):11694-11699.
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421-427.
- Shih RA, Belmonte PL, Zandi PP. A review of the evidence from family, twin and adoption studies for a genetic contribution to adult psychiatric disorders. Int Rev Psychiatry. 2004;16(4):260-283.
- Sklar P, Pato MT, Kirby A, et al. Genome-wide scan in Portuguese Island families identifies 5q31-5q35 as a susceptibility locus for schizophrenia and psychosis. Mol Psychiatry. 2004;9(2):213-218.
- Stahl EA, Breen G, Forstner AJ, et al; eQTLGen Consortium, BIOS Consortium, Bipolar Disorder Working Group of the Psychiatric Genomics Consortium. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet. 2019;51(5):793-803.
- Stanewsky R. Genetic analysis of the circadian system in Drosophila melanogaster and mammals. J Neurobiol. 2003;54(1):111-147.
- Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(10):a006296.
- Thomas DC. Statistical Methods in Genetic Epidemiology. Oxford University Press; 2004.
- Visscher PM, Hill WG, Wray NR. Heritability in the genomics era-concepts and misconceptions. Nat Rev Genet. 2008;9(4):255-266.
- Witte JS, Visscher PM, Wray NR. The contribution of genetic variants to disease depends on the ruler. Nat Rev Genet. 2014;15(11):765-776.
