
1.20 精神医学におけるエピジェネティクス:新しいバイオマーカーと治療法の可能性
アリー・カフマン医学博士
1942年、コンラッド・ワディントンは、「遺伝学(genetics)」と「発生(epigenesis)」(多細胞生物において接合子が異なる細胞型を生み出す発生プログラムを記述するために用いられた古い用語)を融合させ、「エピジェネティクス」という用語を作り出しました。ワディントンによって定義されたエピジェネティクスは、安定した細胞表現型を確立するために、遺伝的形質と環境条件が協力し合うことを表していました。非遺伝的要因が発生において重要な役割を果たすという概念は、「DNA配列の改変を伴わない染色体の変化に起因する、安定して遺伝可能な表現型」としてエピジェネティクスを定義することによってさらに形式化されました。より最近では、「エピジェネティクス」という用語は、環境要因が、遺伝可能であるかどうかにかかわらず、安定した細胞表現型の形成に寄与することを強調するために、さらに広義に解釈されるようになりました。このレビューでは後者の定義を採用し、「エピジェネティクス」という用語は「DNA配列の変化に起因しない、予測可能で安定した、そしておそらく遺伝可能な細胞機能の変化につながる遺伝子発現の変化」を記述するために使用されています。この定義は精神医学において特に重要です。なぜなら、ストレスや薬物乱用などの環境要因が、分裂後のニューロンにおける遺伝子発現を変化させ、それらが遺伝可能ではないものの、大うつ病、不安症、嗜癖などの精神病理に寄与するからです。
表 1.20-1.
遺伝子コードとエピジェネティックコードの差異と類似性
| 変数 | 遺伝子コード | エピジェネティックコード |
| 構成要素 | 4つのヌクレオチド(A, G, C, T)からなる長いDNA鎖 | ヒストン、ヒストン修飾酵素、構造タンパク質、DNA修飾酵素、ATP依存性クロマチン再構築機構 |
| 構造 | 1次元のリニアな鎖 | 動的な3次元構造 |
| 細胞内容 | 全ての細胞で同一 | 各細胞型でユニーク |
| 環境影響 | 環境変化に反応して変化なし | 動的であり、環境的合図や細胞周期の進行に反応して変化する可能性がある |
| 遺伝子発現修飾の可能性 | はい | 環境変化に迅速に反応して変化する。また、長期的に持続する可能性もある |
| 遺伝可能 | はい | 時として |
表1.20-1は、「遺伝子コード」と「エピジェネティックコード」の主要な類似点と相違点のいくつかについて要約しています。構造的に、遺伝子コードは、連結されて長いDNA鎖を形成する1次元のヌクレオチド配列で構成されています。一方、エピジェネティックコードは、DNAをコーティングし、圧縮、解凍、およびループ状にねじれることができる3次元構造に変換する多数のタンパク質および非コードRNAで構成されています。遺伝子コードは体内のすべての細胞で同一ですが、エピジェネティックコードは異なる細胞型間で非常に多様であり、同じ細胞内でも細胞周期の異なる段階で変化する可能性があります。したがって、エピジェネティクスは、同じ遺伝的設計図が異なる細胞型を生成するために使用されるメカニズムを包含します。この概念は図1.20-1に示されており、線形配列の文字として表された同じ遺伝子コードが、基礎となるコード内のどの文字が表示されるかに応じて、2つの異なるエピジェネティックプログラムによって選択的にマスクされ、STAND(上)またはSIT(下)と読み取られます。遺伝子コードは静的であるのに対し、エピジェネティックコードは非常に動的であり、環境的合図に迅速に反応して変化することができます。この動的な特性により、分裂後のニューロンは、シナプス可塑性、学習、行動の変化を駆動する遺伝子の発現を修飾することができます。最後に、遺伝子コードとエピジェネティックコードはいくつかの類似点を共有しています。これらには、世代を超えて伝達され得る方法で遺伝子発現を修飾する能力が含まれます。
エピジェネティクス研究は、精神医学研究において最もエキサイティングで急速に成長している最前線の一つです。これは、6つの特定の領域(表1.20-2に要約)でこの分野を進歩させる可能性を秘めているためです。第一に、エピジェネティクスは、環境要因が精神状態への脆弱性をどのように形成するかを説明する分子メカニズムを提供します。薬物乱用や心的外傷後ストレス障害(PTSD)のような一部のケースでは、環境の寄与は非常に明白です。統合失調症や双極性障害のような他の例では、一卵性双生児間の一致率が50%をわずかに下回るという観察から、強い環境影響を推測できます。本章の目標の1つは、エピジェネティックな変化が特定の環境条件が精神病理のリスクをどのように修飾するかを説明する特定の前臨床および臨床例を提供することです。環境操作も精神症状を緩和するエピジェネティックな変化を誘発しうることを認識することが重要です。実際、抗うつ薬、電気痙攣療法(ECS)、食事、運動など、精神医学における既知の多くの介入は、動物のストレス、快感消失、薬物探索行動への脆弱性を修飾するエピジェネティックな変化を引き起こすことが示されています。エピジェネティクスが環境リスク要因と保護要因をどのように媒介するかをよりよく理解することは、精神疾患を治療するための新しく、そしておそらくより効果的な戦略を生み出す可能性が高いです。
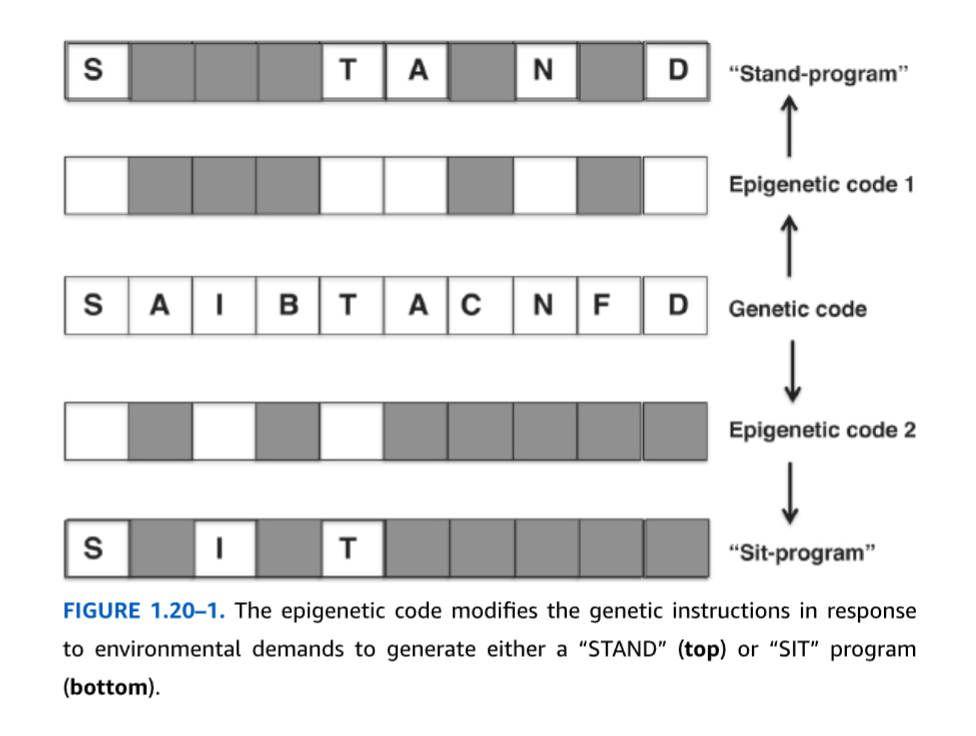
図 1.20-1. エピジェネティックコードは、環境要求に応じて遺伝子命令を修正し、「STAND」(上)または「SIT」プログラム(下)のいずれかを生成します。
第二に、エピジェネティクスは神経発達の誘導において中心的な役割を果たしており、したがって、異常な脳発達が多くの精神状態への脆弱性にどのように寄与するかを説明する可能なメカニズムを提供します。この観察は、エピジェネティックな調節不全によって引き起こされる多数の神経発達障害(表1.20-3)によって裏付けられています。このプロセスのより良い理解は、新しい治療法の開発につながる可能性が高いです(後述の脆弱X症候群のエピジェネティックな病因のセクションを参照)。さらに、本章で要約されているものを含むますます多くの研究が、エピジェネティクスが虐待またはネグレクトに曝された子供たちの脳発達およびいくつかの精神病理への脆弱性を修飾する上で重要な役割を果たすことを示しています(後述で詳細に議論します)。
第三に、エピジェネティクス研究は、**人工多能性幹細胞(iPS細胞)**の生成における最近の進歩に貢献しています。この技術により、研究者は特定の精神疾患(例:統合失調症)を持つ個人から体細胞(例:皮膚細胞)を採取し、それらのエピジェネティックコードを、ニューロン、グリア細胞、さらには完全なオルガノイドに変換するように再プログラムすることができます。これらの細胞はその後、培養して研究することができます。この驚くべきプロセスは、ヒトから機能的な脳組織にアクセスして操作するという、精神医学研究における主要なハードルの1つを解決します。
第四に、過去10年間で、統合失調症(81%)や双極性障害(85%)のような遺伝性の高い精神状態の遺伝的要因をマッピングする上で大きな進歩があったにもかかわらず、遺伝子変異は症例のほんの一部しか説明していません。遺伝率が一卵性双生児と二卵性双生児の一致率の相対的な割合として計算され、一卵性双生児は二卵性双生児よりもはるかに多くのエピジェネティックな類似性を共有するため、エピジェネティクスもこれらの疾患の遺伝性に寄与している可能性があるというコンセンサスが高まっています。さらに、一部のエピジェネティックマーカーが遺伝する能力は、親世代における特定のストレッサーへの曝露が子孫の精神病理のリスクをどのように修飾するかを説明できるかもしれません。例えば、げっ歯類における長期的な親のストレスは、精子における非コード小RNAの内容を変化させ、それが子孫の神経発達、生理機能、行動の変化に関与しています。これらの前臨床的知見は、ストレスを受けた男性に見られる非コード小RNA精子内容の同様の変化を評価するための新しい概念的枠組みを提供します。
第五に、基礎となるDNA配列とそのパッキングクロマチンとの間の動的な相互作用は、遺伝子が特定の環境条件が精神病理を誘発する能力をどのように修飾するかを説明する分子メカニズムを提供します。このような遺伝子-環境相互作用は、セロトニントランスポーターの異なる対立遺伝子と、小児期の虐待が成人期のうつ病のリスクを修飾する能力との間で巧みに実証されており、本章では、遺伝的変化とエピジェネティックな変化がFKBP5の発現をどのように協調させ、PTSD発症のリスクに影響を与えるかを考察することで説明されています。
表 1.20-2.
精神医学におけるエピジェネティクスの可能性
| 貢献 | 実践的な示唆 |
| 1. 環境変化が行動と精神病理をどのように修飾するかを説明する分子メカニズムを提供 | 新規療法 |
| 2. 異常な神経発達が精神病理につながる仕組みを明確化 | より効果的な早期介入 |
| 3. 人工多能性幹細胞の生成を可能にする | 精神疾患を持つ生きた患者から「脳組織」へのアクセスを獲得 |
| 4. 精神病理がどのように遺伝するかを説明する追加の方法を提供 | 新しい診断と介入 |
| 5. 遺伝子と環境要因が相互作用して精神疾患への脆弱性を修飾するメカニズムを解明 | より特異的なバイオマーカーと介入 |
| 6. 精神科の脆弱性を診断するための信頼性の高いバイオマーカーを提供 | 治療反応を診断し評価するための客観的かつ定量可能なマーカーの利用 |
第六に、DNAメチル化など多くのエピジェネティックマーカーは安定したバイオマーカーであり、ごく少量の細胞で正確に定量することができます。これらの特性により、エピジェネティックな変化は、診断および治療反応の評価のための潜在的に価値のあるバイオマーカーとなります。現在、精神医学では、診断または治療反応を評価するための客観的な疾患マーカーが利用できないため、バイオマーカーは切実に必要とされています。ストレス関連遺伝子であるSKA2のメチル化率が自殺行動を予測できることを示唆する最近の知見は、本章の最後のセクションで紹介されています。この種の研究は、精神状態の有用なバイオマーカーを特定する上でのエピジェネティクスの有用性を例示しています。
本章は3つのセクションに分かれています。最初のセクションでは、エピジェネティックな調節に関与する細胞機構について包括的にレビューします。2番目のセクションでは、エピジェネティックな変化が動物の精神医学的に関連する表現型を修飾することが示されたいくつかの前臨床例と、これらの変化を標的とすることで潜在的に新しい精神医学的治療法を生み出す方法を記述します。3番目の最終セクションでは、エピジェネティクスが精神医学における臨床的理解と実践をどのように修飾しうるかを示すいくつかの臨床例を提供します。
エピジェネティック機構
古細菌における進化学的研究は、クロマチンが元々、環境要求に応じて遺伝子発現を迅速に調節するメカニズムとして進化したことを示唆しています。このより複雑な多層的な転写調節の形態は、後に細胞分化をサポートするために適応され、同じ遺伝子コードが異なる細胞型を生成するために使用されます。ゲノムの拡大に伴い、クロマチンは長いDNAを小さな核に収めるために圧縮することが不可欠でした。最後に、クロマチンは、遺伝子コードの完全性を維持するために不可欠な他の多くの機能を果たします。これらには、DNA修復、テロメア形成、セントロメア形成が含まれます。
表 1.20-3.
クロマチン媒介性神経発達障害
| 疾患名 | タンパク質/遺伝子 | クロマチン修飾 | 臨床的特徴 |
| X連鎖αサラセミア/精神遅滞症候群 (ATR-X) | ATRX/XH2, SWI/SNFファミリータンパク質の一員(クロマチンリモデリング酵素) | α-グロビン遺伝子座のダウンレギュレーションに関与する欠陥クロマチンリモデリング | X連鎖遺伝;精神遅滞、溶血性貧血、脾腫、顔面、骨格、生殖器の異常 |
| プラダー・ウィリー症候群 | 親由来15q11-13染色体が開いたクロマチン状態を維持し、正常な発達に必要な遺伝子の発現を可能にする | 親由来15q11-13遺伝子座の欠失 | 初期の新生児期における低緊張と摂食障害に続き、過食症、小児肥満、性腺機能低下症、軽度の知的障害 |
| アンジェルマン症候群 | 母由来15q11-13遺伝子座のクロマチンが高度に凝縮し、UBE-3AアンチセンスRNAの発現を妨げ、UBE3A遺伝子の発現を可能にする | 母由来15q11-13遺伝子座の欠失または母由来UBE3A遺伝子の変異 | 大脳萎縮、小脳の異常な髄鞘形成、発作、精神遅滞 |
| コフィン-ローリー症候群 | RSK2(キナーゼ) | 異常なヒストンアセチル化およびリン酸化 | X連鎖遺伝;精神運動遅滞、骨格および頭蓋顔面の変形 |
| 脆弱X症候群 | FMR1およびFMR2 | FMR1およびFMR2プロモーターにおける拡張された三ヌクレオチド反復のDNA高メチル化 | 最も一般的な遺伝性知的障害、自閉症行動の兆候、大きな耳と巨睾丸を伴う長く細い顔 |
| 免疫不全-セントロメア不安定性-顔面異常症候群 (ICF) | Dnmt3B(新規DNAメチル転移酵素) | 1番、9番、16番染色体のセントロメア領域の低メチル化 | 軽度の精神遅滞、免疫不全、顔面異常 |
| ルビンシュタイン・テイビ症候群 | CBP(HAT) | ヒストンアセチル化の減少、常染色体優性遺伝 | 精神遅滞、異常な顔面特徴、成長阻害 |
| レット症候群 | MeCP2(MBP) | メチル化DNAへのMeCP2の異常なリクルート | X連鎖、広汎性発達障害、主に女児に影響し、脳発達の停止、認知機能低下、自閉症様行動を伴う |
| 9q34末端欠失症候群 | GLP(HMT) | 異常なH3K9メチル化 | 重度の精神遅滞、低緊張、顔面異常 |
クロマチンの基本的な折りたたみ単位はヌクレオソームです。ヌクレオソームは、DNAループ(図1.20-2A)によって巻き付かれたヒストン八量体コアで構成されています。八量体コアは、高度に保存された、正電荷を帯びた4つのヒストンタンパク質(H2A、H2B、H3、H4)から組み立てられます。H3とH4は四量体を形成し、2つのH2A/H2Bダイマーの間に挟まれて八量体コアを形成します。八量体コアは、負電荷を帯びたDNAとヒストンに見られる正電荷を帯びたアミノ酸との間に形成される多数のイオン結合によって安定化された146bpのDNAループによって巻き付かれています。各ヒストンタンパク質は、突出して周囲の空間を探る長い無構造のN末端テールを持っています。このテールは、クロマチンが凝縮または開放する能力において重要な役割を果たします。
「ビーズ・オン・ア・ストリング(紐につないだ真珠)」としても知られる開いたクロマチン構造では、隣接するヌクレオソームは短い(10〜80bp)リンカーDNAによって分離されています(図1.20-2B)。「ビーズ・オン・ア・ストリング」クロマチン構造は、基礎となるDNAの特性を2つの重要な方法で変化させます。第一に、DNAを約5〜10倍に圧縮します。第二に、巻き付いたDNAの大部分は、転写活性化因子の結合にアクセスできません。その結果、最もアクセスしやすい形態であっても、クロマチンは基礎転写の障壁となります。さらに、ヒストンの長いN末端テールは、隣接するヌクレオソーム間の接触を作り、クロマチンを凝縮させる傾向があります。これらの高次フィラメントは、5番目のヒストンであるヒストンH1の結合によって安定化され、さらにDNAを圧縮し、アクセスしにくくする高次30nmフィラメントを形成します。クロマチンがアクセスしにくい、よりコンパクトな構造に巻き付くこの自然な傾向は、ヒストンN末端テールの翻訳後修飾、クロマチンリモデリング機構のリクルート、および不変ヒストンの使用を含むいくつかのメカニズムによって調節されます(後述で詳細に記述)。要約すると、ヌクレオソーム単位は、DNAの化学的性質や遺伝子コードを変化させることなく、遺伝子発現に不可欠なDNA配列へのアクセスを調節します。
ヒストンの修飾
ヒストンのN末端テールは、アセチル化、メチル化、リン酸化、ユビキチン化、スモイル化などの主要な例として、多数の翻訳後修飾によって修飾されています。これらの修飾は可逆的であり、環境的合図に応じてクロマチンを凝縮または開放するように活動を協調させる大きな多サブユニットタンパク質複合体によって実行されます。ヒストン修飾は、3つの異なるメカニズムを介してクロマチン構造に影響を与えます。第一に、一部の修飾は、ヒストンテール上の正電荷を帯びた残基と負電荷を帯びたDNA骨格との間のイオン結合を破壊し、DNA-ヒストン結合を緩めます。第二に、ヌクレオソームは、そのN末端テールを介して互いに接触します。これらのヌクレオソーム間接触は、翻訳後修飾によって強化または破壊されることがあります。第三に、ヒストンテールの変化は、クロマチンリモデリング機構をリクルートする結合部位を提供します。これらのタンパク質は、ATP加水分解を利用して、ヌクレオソームをクロマチンからスライドまたは除去することができます(図1.20-3)。
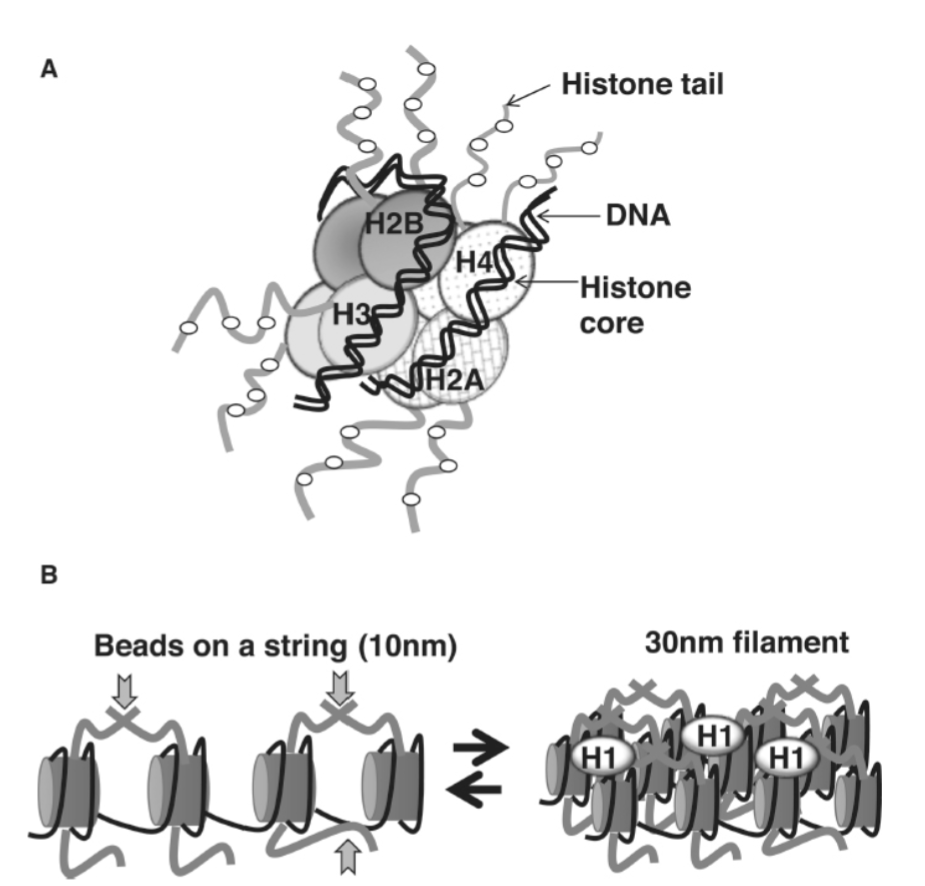
図 1.20-2. A: ヌクレオソームはクロマチンの基本的な折りたたみ単位です。B: クロマチンは、「ビーズ・オン・ア・ストリング(紐につないだ真珠)」状の10nmフィラメントから、ヒストンH1(薄い楕円)の存在によって安定化された、よりコンパクトな30nmフィラメント構造へと移行することができます。ヒストンテール(灰色)は、隣接するヌクレオソーム間(上向き矢印)および基礎となるDNA(下向き矢印)と接触します。
ヒストンのアセチル化
アセチル化はリジン残基を中和し、ヒストンのN末端テールの正電荷を減少させます。この正電荷の減少は、負電荷を帯びたDNAや隣接するヌクレオソームとの接触を阻害すると考えられています。ヒストンのアセチル化は、転写活性化や、DNA複製や修復など、基礎となるDNAへのアクセスを必要とする他の細胞プロセスと関連しています(表1.20-4)。ヒストンアセチルトランスフェラーゼ(HAT)はヒストンH3とH4のテールにあるリジン残基をアセチル化し、ヒストンデアセチラーゼ(HDAC)はこれらのアセチル化を除去します(図1.20-4)。アセチル化は、ヒストン3(例:9、14、18、23)およびヒストン4(例:5、8、12、16)に体系的に配置されたリジン(K)を修飾します。異なるHATとHDACが特定の残基のアセチル化と脱アセチル化を担当しています。例えば、ヒストンH3では、K14とK18はcAMP応答エレメント結合タンパク質(CREB)結合タンパク質(CBP/P300)によってアセチル化され、K9、K14、K18は異なるHATであるPCAF/GCN5によってアセチル化されます。この酵素特異性は、このシステムに作用するより選択的で標的を絞った薬物療法を開発する機会を提供します。
再構築された精製ヒストンを用いた最近の研究では、ヒストン4のリジン14の単一のアセチル化(H4K14-Ac)が、隣接するヌクレオソーム間の相互作用を妨げることによって、クロマチンを「ビーズ・オン・ア・ストリング」状態に維持するのに十分であることがわかりました。興味深いことに、H4K14-Ac修飾は、クロマチンリモデリング酵素ACFがヌクレオソームをスライドさせる能力を阻害し、この修飾がクロマチンをオープンな状態に「ロックする」ことを示唆しています。
ヒストンのアセチル化がクロマチンを開放することと一致して、ヒストンの脱アセチル化はクロマチン凝縮と転写抑制につながります(図1.20-4)。HDACは、クラスI、クラスII、およびNAD依存性クラスIIIのSIRファミリーの3つのクラスに分類されます。これらのクラスは構造的な類似性に基づいており、したがって異なる薬理学的薬剤によって阻害されます。一般に、HDACは脱アセチル化するリジン残基に関してHATよりも特異性が低いようです。異なるHDACは、核内の異なる区画を標的とし、結合することで、ある程度異なる機能を果たすことができます。例えば、HDAC1はクロマチン凝縮と抑制に一般的な役割を果たすようですが、HDAC3は核周辺部に繋留されており、そこで高度に抑制的なヌルクロマチンを形成します(後述の「クロマチンドメイン」のセクションを参照)。
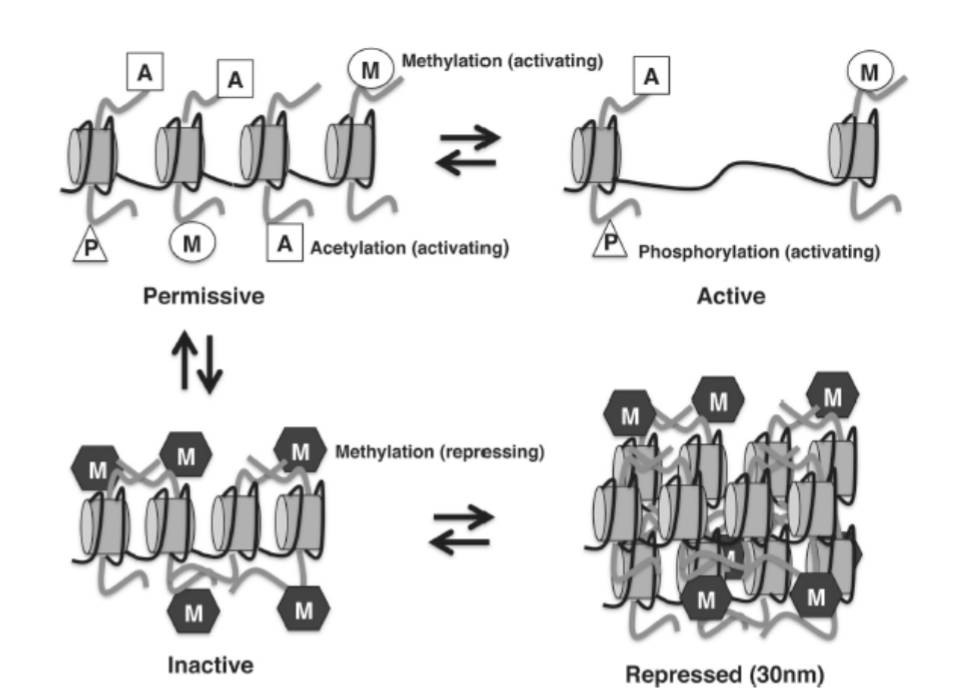
図 1.20-3. 「許容的クロマチン」(左上)はアセチル化(灰色の四角)され、メチル化(開いた楕円)され、リン酸化(灰色の三角形)されています。これらのヒストン修飾は、ヌクレオソーム間およびDNA-ヌクレオソーム間の接触を阻害することによってクロマチンを開放し、ATP依存性クロマチンリモデリング酵素(図示せず)をリクルートしてさらにクロマチンを開放し、「活性クロマチン」(右上)を形成します。許容的クロマチンは、活性化ヒストン修飾を除去し、抑制性ヒストンメチル化(窓枠模様の六角形)を追加することによって、「不活性クロマチン」(左下)に修飾され得ます。これらの抑制性修飾は、ヌクレオソーム間およびDNA-ヌクレオソーム間の接触を促進することによってクロマチンを凝縮させます。不活性クロマチンは、ATP依存性クロマチンリモデリング酵素やその他の構造タンパク質(図示せず)をリクルートすることによって、さらに凝縮して「抑制クロマチン」(右下)を形成することができます。
表 1.20-4.
クロマチンを修飾する酵素活性
| 修飾の種類 | 修飾される残基 | クロマチンへの影響 | 修飾酵素 | 脱修飾酵素 |
| ヒストンのアセチル化 | H3K9, H3K14, H3K18, H3K23, H4K5, H4K8, H4K12, H4K16 | クロマチン開放/転写活性化 | HAT (GCN5, SRC-1, CBP) | HDAC (HDAC1, HDAC2, HDAC5) |
| ヒストンのメチル化 (活性化) | H3K4, H3K36, H3K79 | クロマチン開放/転写活性化 | HMT (Set7/9, MLL, SUV39H/H2) | HDMT (LSD1, JMJD2A) |
| ヒストンのメチル化 (抑制) | H3K9, H3K27, H4K20 | クロマチン凝縮/転写抑制 | HMT (GLP, G9A, EZH2) | HDMT (KDM3A/JMJD1A, JMJD2A, JMJD3) |
| ヒストンのリン酸化 | H3S10, H3S28 | クロマチン開放/転写活性化 (初期応答遺伝子) | HK (AURORA/IP1, MSK1/MSK2) | HP (DUSP1) |
| クロマチンリモデリング酵素 | 該当なし | クロマチン開放または凝縮 | 該当なし | 該当なし |
| DNAメチル化 | CpGジヌクレオチド内のシチジン | クロマチン凝縮/転写抑制 (主に) | DNMT1, DMT3a, DNTM3b | Tetファミリー, Gadd45ファミリー, AID, SMUG1/TDG |
ブロモドメイン構造を持つタンパク質は、アセチル化されたヒストンに結合してオープンクロマチンを維持することができます。CBPのHATドメインの変異は、精神遅滞と心臓の異常を伴うルビンシュタイン・テイビ症候群を引き起こします(表1.20-3)。興味深いことに、この疾患のマウスモデルでは、CBPの低レベルに関連する認知機能の欠損が、成体マウスにHDAC2阻害剤を投与することで回復することが示されました。これらの知見は、分裂後のニューロンにおけるHATとHDACの相対的な活性が、神経発達障害、老化、薬物依存症に広範な影響を与える神経可塑性において重要な役割を果たすことを示唆しています(「前臨床研究におけるエピジェネティクス」を参照)。実際、HDAC2の増加は、学習と記憶に必要な遺伝子をサイレンシングすることが示されており、HDAC2の活性はアルツハイマー病で上昇していることが判明しています。現在、特定のHDAC阻害剤が、早期アルツハイマー病、統合失調症、外傷性脳損傷などの状態における認知機能を改善するために使用できるかをテストする臨床研究が進行中です。

逐語的に正確に日本語に翻訳します。
図 1.20-4. ヒストンテールは特定の残基で可逆的に修飾されます。HAT – ヒストンアセチルトランスフェラーゼ、HDAC – ヒストンデアセチラーゼ、HMT – ヒストンメチルトランスフェラーゼ、HDMT – ヒストン脱メチルトランスフェラーゼ、HK – ヒストンキナーゼ、HP – ヒストンホスファターゼ。
ヒストンのメチル化
ヒストンのメチル化は、リジン(K)とアルギニン(R)の両方がメチル化され得るため、アセチル化よりも複雑です。さらに、各リジンは1回、2回、または3回メチル化される可能性があり、これがさらなる複雑さを生み出しています。リジンメチルトランスフェラーゼは、アセチル化酵素と比較してより選択的であり、ヒストンテール内の特定のリジン残基を標的とします。3つの部位でのメチル化は転写活性化(H3K4、H3K36、H3K79)と関連付けられていますが、他の3つのリジン残基でのメチル化は抑制(H3K9、H3K27、H4K20)と関連付けられています(要約は表1.20-3を参照)。転写調節に加えて、ヒストンのメチル化はDNA修復にも役割を果たします。
他のクロマチン修飾と同様に、メチル化されたリジンはヒストン脱メチル化酵素によって脱メチル化され、動的で可逆的なクロマチン変化を可能にします(図1.20-4)。クロモドメイン(例:クロモ、チューダー、MBT)または構造的に無関係なPHDモチーフを含むタンパク質は、メチル化されたヒストンに結合することができます。このようなタンパク質は、クロマチンリモデリング酵素や他のヒストン修飾酵素を含む大きな複合体を形成します。これらの複合体はメチル化されたヒストンにリクルートされ、複合体の正確な組成に応じて、クロマチンを効率的に開いたり閉じたりすることができます。例えば、HOXC8遺伝子の適切な活性化は、すべての脊椎動物における正常な軸発達に不可欠です。HOXC8遺伝子の活性化には、発生の特定の期間におけるヒストンメチル化が必要です。このメチル化ステップは、ブロモドメインPHDフィンガートランスクリプションファクター(BPTF)を介してヌクレオソームリモデリング因子(NURF)をリクルートするために必要です。NURF複合体には、HOXC8プロモーターからヌクレオソームを除去し、転写活性化を可能にするクロマチンリモデリング酵素(SNF2L)が含まれています。BPTFがメチル化されたプロモーターに結合する能力を妨げる変異は、おたまじゃくしにおけるHOXC8の異常発現と異常な軸形成を引き起こします。この一連の出来事は、ヒストン修飾が追加の機構をリクルートし、クロマチンをよりアクセスしやすくし、正常な軸発達に必要な遺伝子の発現を可能にするという、多層的なクロマチン調節の協調を示しています(図1.20-5)。
ヒストン3のリジン9のメチル化(H3K9-me2/H3K9-me3)は、セントロメア周辺/構成的ヘテロクロマチンとして知られる特殊なクロマチン形態を維持する上で重要な役割を果たします。セントロメア周辺クロマチンの主な機能の1つは、細胞分裂中の染色体の整列と分離に使用されるセントロメアの形成をサポートすることです(表1.20-4も参照)。H3K9は特定のヒストンメチルトランスフェラーゼ(Suv39h1/h2)によってメチル化され、それがそのクロモドメインを介してリプレッサータンパク質HP1の結合を誘発します。HP1はその後、いくつかの他のタンパク質と結合して複合体を形成し、基礎となるDNAの大部分にわたってこの特殊な抑制されたクロマチン構造を拡張させます。これは、HP1が追加のSuv39h1/h2をリクルートする能力によって達成され、さらなるヒストンメチル化とHP1のリクルートがDNAを介して伝播することを可能にします(図1.20-6)。HP1はまた、DNAメチルトランスフェラーゼであるDNMT1とDNMT3aとも関連しており、DNAメチル化とさらなるクロマチン凝縮を可能にします。最後に、複製中、HP1は複製フォークにヒストンを堆積させるクロマチン因子1(CAF1)タンパク質と関連しています。複製フォークにおけるHP1、Suv39h1/h2、DNMT1、CAF1を含む複合体の存在は、セントロメア周辺クロマチンが娘細胞に遺伝する可能性のあるメカニズムと、DNAメチル化とヒストン修飾活性がどのように協調するかを示す良い例を提供します(後述の「DNAメチル化」セクションを参照)。
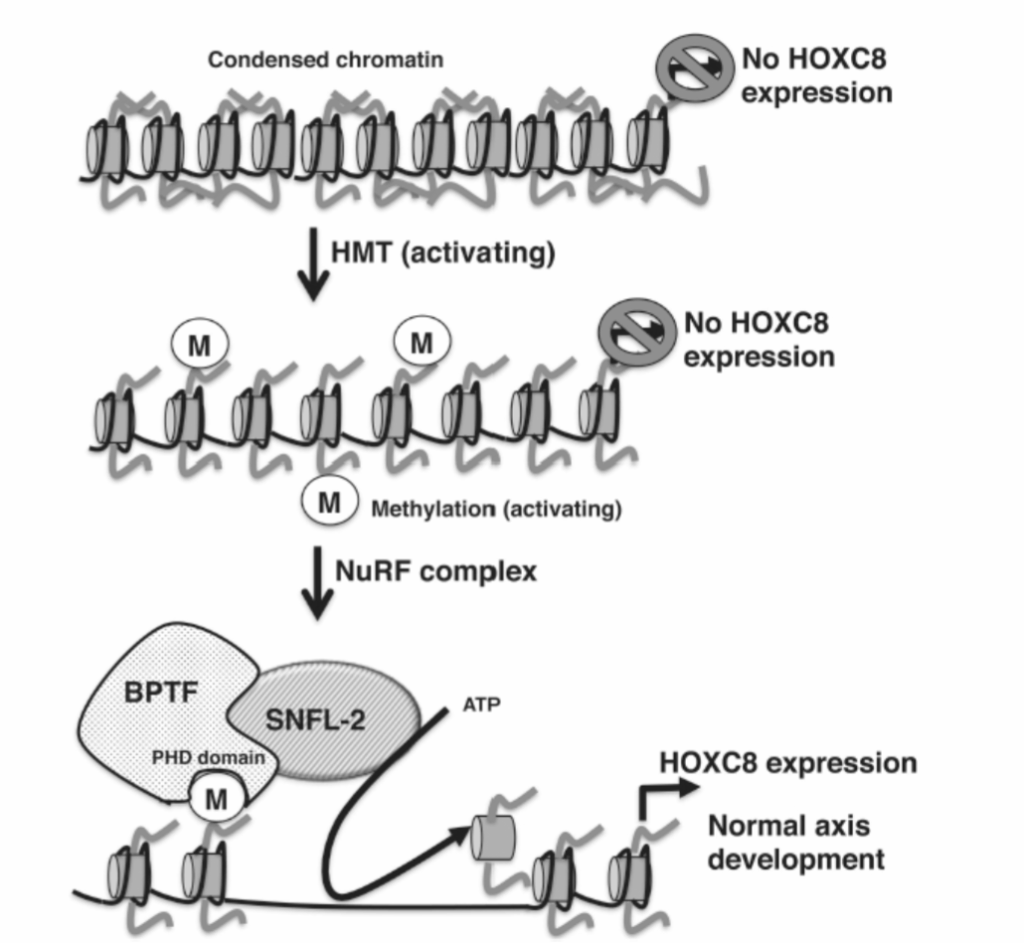
図 1.20-5. ヒストンメチル化は、そのPHDドメインを介してBPTFをリクルートし、NuRF複合体の一部としてクロマチンリモデリング酵素SNFL2を連れてきます。SNFL2の結合により、プロモーターからATP依存的にヌクレオソームが除去され、HOXC8の発現と脊椎動物の正常な軸発達が起こります。
ヒストンのメチル化は、抑制性ポリコームクロマチン構造の形成にも不可欠です。これは、胚性幹細胞の多能性維持、細胞周期の進行、細胞分化、およびX染色体不活性化において重要な役割を果たします。このプロセスでは、H3K27でのヒストンメチル化は、高度に進化的に保存されたタンパク質複合体であるポリコーム抑制複合体(PRC2)によって触媒されます。PRC2は、他のDNA結合タンパク質や非コードRNAとの相互作用を介して、DNAの特定の領域にリクルートされます。リクルートされると、PRC2はH3K27をメチル化して、ジメチル化状態およびトリメチル化状態(H3K27-me3)を形成します。これらは複合体全体のテザリング部位として機能します。H3K27-me3は、クロマチンリモデリング機構(PRC1)もリクルートし、クロマチンをさらに凝縮させて、高度に抑制的なポリコーム構造を形成します(図1.20-7および表1.20-4を参照)。興味深いことに、PRC2複合体はDNA合成中もDNAへの結合を維持しており、これによりH3K27-me3およびポリコームクロマチン抑制が娘細胞で維持されるメカニズムが提供されます。PRC2のヒストンメチラーゼ活性における変異は、ポリコーム抑制の娘細胞への安定した伝播を妨げ、X不活性化や細胞の多能性から分化への移行を含む多くの発達プログラムにおいて、このエピジェネティックな遺伝形式におけるメチル化の本質的な役割を示唆しています。
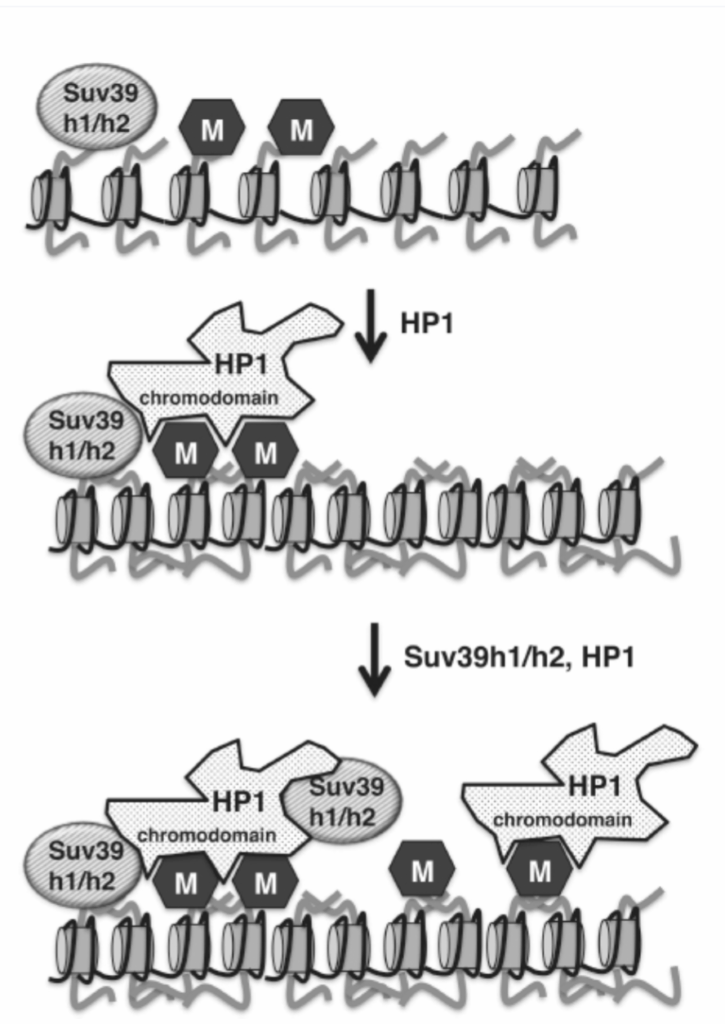
図 1.20-6. HMT Suv39h1/h2がH3K9をメチル化し(上)、そのクロモドメインを介してHP1をリクルートします(中)。HP1はSuv39h1/h2に結合し、この抑制的なセントロメア周辺/構成的ヘテロクロマチンの伝播を可能にします(下)。
ヒストンのリン酸化
ヒストンのリン酸化は、有糸分裂中の染色体分離、初期応答遺伝子の誘導、およびDNA修復において役割を果たします。オーロラ/Ip1や有糸分裂促進因子およびストレス活性化プロテインキナーゼ1および2(MSK1/MSK2)などのヒストンキナーゼは、ヒストン3のセリン(S)10および28をリン酸化します。これらのセリン残基がリジン9および27に近接していることは、これらのリジンがアセチル化されるかメチル化されるかという決定に影響を与えます。リジン9および27のアセチル化は転写を活性化する傾向があるのに対し、同じ部位でのメチル化は転写を抑制するため、この影響は重要です(図1.20-4)。ストレス、コカイン、神経伝達の変化、紫外線などのいくつかの環境刺激は、MSK1およびMSK2を活性化することによりH3S10のリン酸化を誘発します。活性化されたMSK1は、14-3-3タンパク質、クロマチンリモデリング機構、およびHATを含む複合体として、c-fosやJunなどの初期応答遺伝子のプロモーターにリクルートされます。この複合体は、細胞の種類と環境に応じて適切な細胞応答を提供する初期応答遺伝子の発現を効率的に活性化します。歯状回では、この応答が無力感行動を促進する一方、線条体では、この応答が薬物探索行動を修飾します(後述の「前臨床研究におけるエピジェネティクス」セクションを参照)。
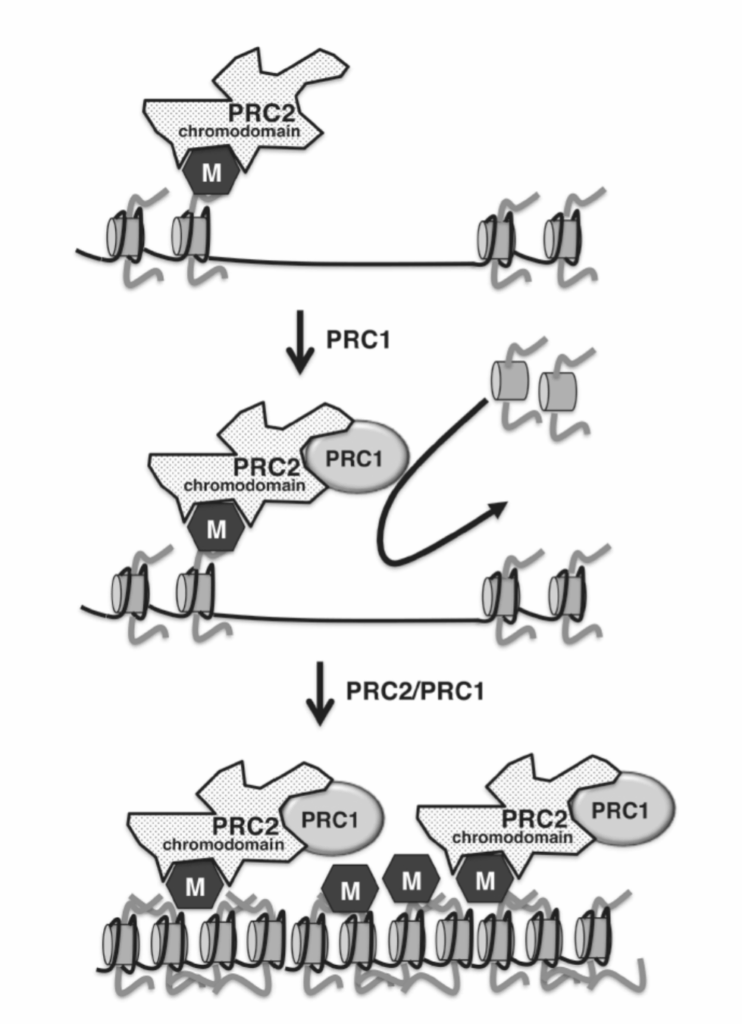
図 1.20-7. PRC2複合体はH3K27-me3を生成するHMT活性を持ち、これがPRC2のクロマチンへの結合をさらに安定化させます(上)。PRC2はPRC1複合体をリクルートし、ヌクレオソーム密度を増加させることによってクロマチンをさらに凝縮させます(中)。これらの新たにリクルートされたヌクレオソームはPRC2によってメチル化され、このポリコーム抑制クロマチンの伝播を可能にします(下)。
ATP依存性クロマチンリモデリング
ヌクレオソームは、活発に転写されているハウスキーピング遺伝子に沿って、決まったパターンで分布しています。このパターンは酵母から哺乳類まで高度に保存されており、ハウスキーピング遺伝子のプロモーター領域において、250bpのヌクレオソームフリー領域(NFR)が2つの配置されたヌクレオソームに挟まれていることが特徴です(図1.20-8)。同様のNFRは、遺伝子の3’末端およびエンハンサー領域にも見られます。このパターンにより、転写機構が結合し、ハウスキーピング遺伝子を構成的に発現させることができます。
ヌクレオソームの配置は、生体内では2つのメカニズムによって達成されます。第一に、プロモーター領域内の特定のDNA配列がヌクレオソームの位置を誘導することができ、これは遺伝子コードがエピジェネティックコードに影響を与える重要な例を提供します。しかし、この配置は静的であり、環境要求に応じてクロマチンが迅速に再編成されることを許しません。
第二に、そしておそらくより重要なメカニズムは、クロマチンリモデリング酵素によって媒介されます。これらの酵素は、クロマチン結合モチーフを備え、ATPを使用してヌクレオソームをDNA鎖に沿って除去、追加、またはスライドさせます。場合によっては、クロマチンリモデリング酵素は、特定のプロモーター配列からヌクレオソームを除去することによってNFRを維持します。これにより、転写因子がDNAに結合して転写を活性化することができます(図1.20-8)。あるいは、ヌクレオソームはクロマチンリモデリング酵素によってNFRに押し込まれ、そこで転写をブロックし、高次クロマチン構造を形成することができます(図1.20-8)。クロマチンリモデリング酵素のリクルートは、マウスにおける慢性的なコカイン曝露後の依存症様行動の発症に関与していることが示されています。これは、サイクリン依存性キナーゼ5(CDK5)などの後期発症型依存症関連遺伝子のプロモーター修飾を介しています(「コカイン依存症のエピジェネティックメカニズム」のセクションを参照)。
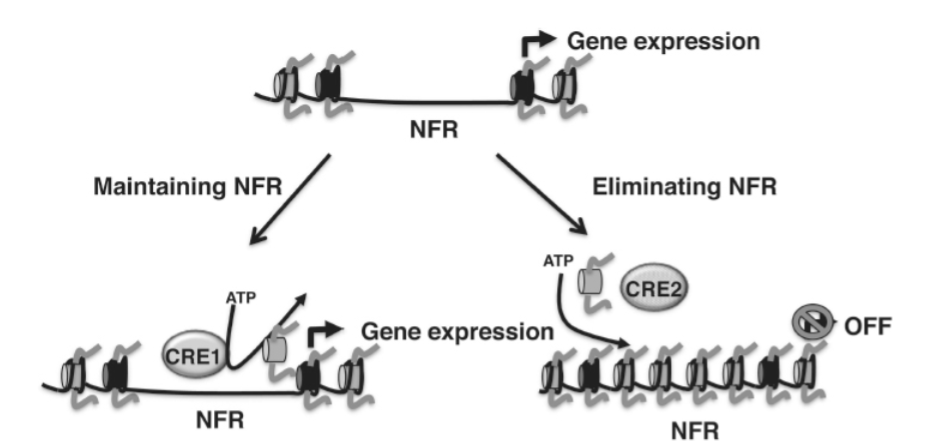
図 1.20-8. 上: ヌクレオソームフリー領域 (NFR) は、NFRの両側にヌクレオソームを配置するDNA配列 (濃い灰色で示される) によって受動的に維持され得ます。これにより、転写因子が結合して遺伝子発現を活性化することができます。多くの場合、NFRはヌクレオソームのATP依存性除去 (左下のパネルのCRE1として示される) によって積極的に維持されます。ATPリモデリング活性 (右下のパネルのCRE2) は、ATPを使用してヌクレオソームを配置し、NFRをアクセス不能にして、転写をオフにします。
DNAメチル化
遺伝子コードの化学構造を変化させないヒストン修飾とは異なり、DNAメチル化はシトシンの分子構造を直接変化させます。これは、ドナー分子からシトシン環の5’位にメチル基を転移させて**5-メチルシチジン(5mc)**を生成することによって達成されます。脊椎動物では、**DNAメチルトランスフェラーゼ(DNMT)**酵素が、ジヌクレオチド(CpG)パリンドローム配列内のシトシンをメチル化します。シトシンメチル化は、DNAの逆平行鎖上のグアノシンと正しいワトソン-クリック塩基対を形成する能力には影響しないため、基礎となる遺伝情報には影響しません。代わりに、DNAメチル化は、遺伝可能な方法でDNAの特定の場所をマークし、ヒストンおよびヌクレオソームリモデリング機構がクロマチンを開放または圧縮する「重労働」を行うことを可能にします。
哺乳類ゲノム内では、CpGジヌクレオチドは、CpGアイランド(CGI)として知られる高密度領域にクラスター化されているか、ゲノム全体に散発的に見られます。CGIは通常、活性なハウスキーピング遺伝子のプロモーターをマークしており、したがって一般的に非メチル化されています。稀なケースでは、CGIがメチル化されると、遺伝子発現を阻害し、多くの場合遺伝する高度に抑制的なクロマチン構造を形成します。いくつかのよく知られた例には、X染色体不活性化やインプリンティング遺伝子が含まれます。インプリンティング遺伝子の異常な調節がいくつかの形態の知的障害につながるという観察は、正常な神経発達のためのこれらのCGIの正確なメチル化の重要性を示しています(表1.20-3および後述のプラダー・ウィリー症候群の救済のセクションを参照)。
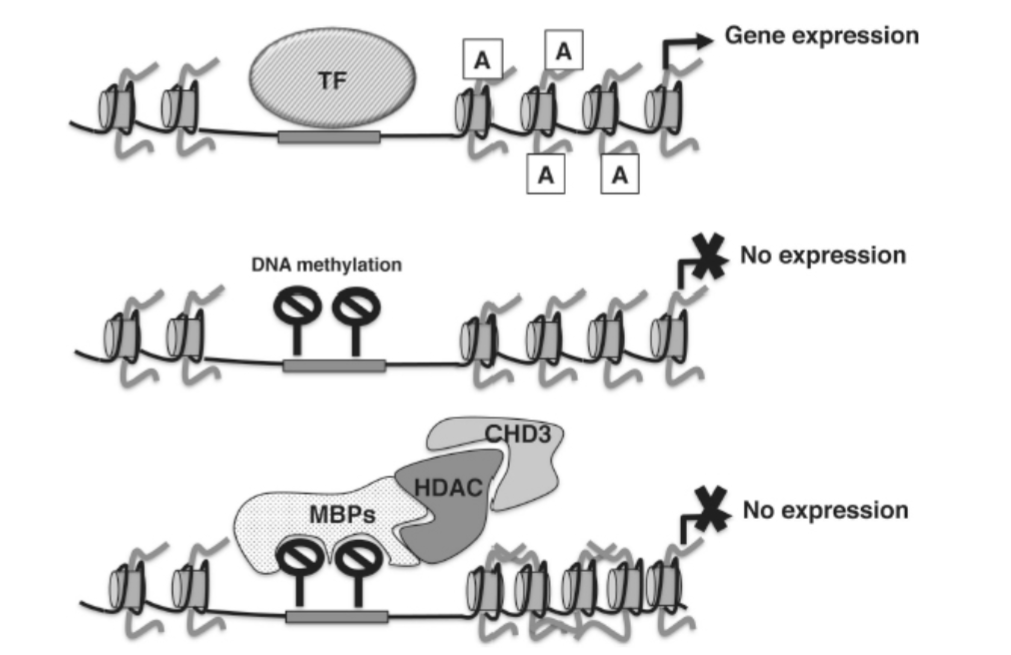
図 1.20-9. 上: DNAメチル化がない場合、一般的な転写因子 (TF) がプロモーター内の特定のDNA配列に結合し、遺伝子発現を活性化します。中: DNAメチル化 (黒い進入禁止標識) は、TFがその結合部位に結合するのを妨げ、したがって遺伝子発現を妨げます。下: NuRD複合体は、メチル結合タンパク質 (MBP) を介してメチル化されたDNAにリクルートされます。これらのMBPは、HDACやクロマチンリモデリングタンパク質CHD3などの酵素活性と複合体を形成し、DNAを圧縮して遺伝子発現を停止させます。
散発的な領域におけるCpGのメチル化は、通常、遺伝子発現を下方制御します。この下方制御は、次の2つの方法のいずれかで達成されます。第一に、DNAメチル化は、転写因子がその結合部位に結合するのを妨げることができます(図1.20-9)。第二のメカニズムは、メチル結合タンパク質(MBP)がメチル化されたシトシンに直接結合することを含みます。MBPがメチル化されたDNAにドッキングすると、クロマチンをさらに圧縮し、遺伝子発現を阻害する酵素がリクルートされます。この抑制カスケードの良い例は、ヌクレオソームリモデリング脱アセチル化酵素(NuRD)抑制複合体です。NuRDは、クロマチンを相乗的に抑制する高度に保存された多サブユニット複合体です。NuRD複合体には、2つのHDAC(HDAC1とHDAC2)、クロマチンリモデリングサブユニット(CHD3またはCHD4)、およびいくつかのMBP(MBD2とMBD3)が含まれます。MBPはNuRD複合体をメチル化されたシトシンにリクルートし、そこでCHD3/CHD4を介したATP加水分解がヌクレオソームを圧縮し、HDACによる脱アセチル化を促進します。ATP加水分解とヒストン脱アセチル化との間のこの相乗作用は、クロマチンリモデリング酵素がヒストンテールを脱アセチル化によりアクセス可能にする能力によるものと考えられています。その結果、メチル化された部位の周りに高度に抑制的なクロマチン構造が形成されます(図1.20-9)。したがって、DNAメチル化は、遺伝子コード(すなわち、特定の結合部位内のCpG)と他のクロマチン修飾機構(例:図1.20-9、1.20-13、および1.20-14)との間の**「分子的な橋渡し」**と考えることができます。本章の臨床セクションでは、この概念の例として、遺伝子コードと初期の人生ストレスによって誘発されるDNAメチル化の変化がどのように相互作用してFKBP5遺伝子の発現を修飾し、その後のPTSDへの脆弱性を変化させるかを示しています。
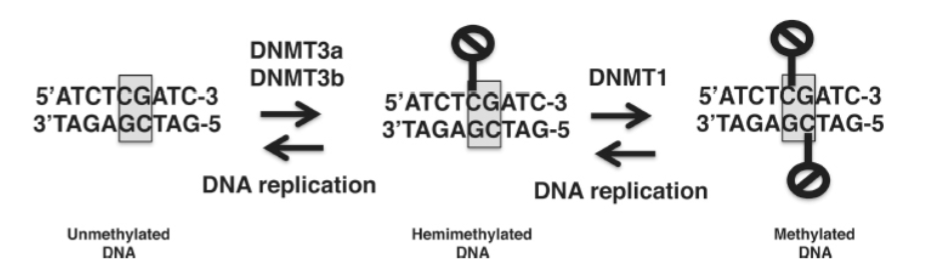
図 1.20-10. 上: 非メチル化DNAは、de novo DNMTであるDNMT3aとDNMT3bによってメチル化され、ヘミメチル化DNAを形成します。ヘミメチル化DNAは、維持DNMTであるDNMT1によってさらにメチル化されます。DNAメチル化は、DNA複製の各ラウンド後に失われます。CpGジヌクレオチドは灰色で示され、DNAメチル化は黒い「進入禁止」標識で示されています。
2つのde novo DNMT酵素(DNMT3aおよびDNMT3b)は、裸のDNAまたは非メチル化DNA上のCpG配列をメチル化できますが、維持DNMT(DNMT1)はヘミメチル化配列のみをメチル化します(図1.20-10)。de novo DNMTは、正常な発達中に発生する2つのグローバルな脱メチル化イベントの後にクロマチンを再メチル化する役割を担っています。最初のグローバルな脱メチル化は胚の配偶子で起こり、これらの細胞を精子または卵子に再プログラムするために行われます。2番目のグローバルな脱メチル化は受精直後に起こり、接合子に多能性状態を確立するために必要です。DNMT3aまたはDNMT3bのいずれかの欠失は胚致死であり、正常な発達におけるde novoメチル化の重要性を強調しています。de novo DNMTは分裂後のニューロンでも発現しており、そこでシナプス可塑性、学習、コカインへの応答に関与する遺伝子の発現を調節するプロモーター要素をメチル化します。
2つのde novo DNMT(DNMT3a、DNMT3bなど)とは対照的に、DNMT1は裸のDNA(すなわち非メチル化DNA)を効率的にメチル化しません。代わりに、DNMT1は、片方の鎖のみがメチル化されたDNAを効率的にメチル化します(図1.20-10)。このヘミメチル化DNAは、完全にメチル化されたCpG部位のDNA複製後に一般的に形成されます。複製フォークにおけるDNMT1の存在により、母鎖に存在するDNAメチル化パターンを新しく形成された娘鎖に忠実にコピーすることができます。このメカニズムは、有糸分裂におけるDNAメチル化の遺伝性を担っています。神経幹細胞(NSCs)におけるDNMT1の欠失は、胚発生中のグリア形成(すなわち、グリア細胞の産生)の早期誘導と、神経分化およびシナプス形成における複数の欠陥を引き起こします。さらに、げっ歯類を早期の人生ストレスに曝露すると、NSCsにおけるDNMT1の発現が増加し、それがレチノイン酸受容体のプロモーター領域のメチル化増加と関連しています。このプロモーターメチル化は、レチノイン酸受容体の発現を減少させ、これによりNCSsがニューロンに分化する能力に欠陥が生じます。要約すると、NSCsの正常な分化には適切なDNMT1活性が必要です。
長年の支配的なドグマは、DNAメチル化は、DNMT1の不活性化と結合したDNA複製(図1.20-10参照)によってのみ除去できる安定したエピジェネティックマーカーである、というものでした。このプロセスは受動的脱メチル化として知られています。したがって、分裂後のニューロンにおけるDNAメチル化の存在は、遺伝子発現の長期抑制を引き起こす安定したマーカーであると考えられていました。この主張は、メチル基とシトシン環の間に形成される炭素-炭素共有結合が非常に安定であり、in vivoでこの結合を切断することが決定的に示された酵素活性がこれまでないという観察に基づいています。しかし、ますます多くの研究が、一部のCpG部位の脱メチル化が分裂後のニューロンで迅速に起こりうることを一貫して示しています。例えば、海馬の歯状回にある分裂後のニューロンで調べられた合計219,000のCpG部位のうち、単回ECS投与後4時間で約3000の部位でDNAメチル化の変化が示されました。これらの部位の約3分の2はDNAメチル化の増加を示し、残りの3分の1はDNAメチル化の減少を示し、迅速かつ活性な脱メチル化を示しています。DNAメチル化および脱メチル化は、シナプス形成に関与する遺伝子の発現を修飾し、ECSが海馬における神経可塑性をどのように修飾するかを説明する可能なメカニズムを提供します。興味深いことに、3日間の自発的な運動後にも同様のDNAメチル化の変化が観察されており、ECSと運動が海馬におけるシナプス形成を修飾するために類似のエピジェネティックメカニズムを共有していることを示唆しています。
ニューロンにおいてDNAが迅速に脱メチル化され得るという観察は、その責任メカニズムの集中的な探索につながりました。異なるモデルが提案されており、いくつかの経路が関与している可能性を考えると、この問題をさらに明確にするためには追加の研究が必要です。それにもかかわらず、このプロセスにおけるTen-Eleven Translocation(TET)ファミリータンパク質、特にTet1の役割を支持する説得力のある証拠が出現しています。このFe(II)/2-オキソグルタル酸依存性ジオキシゲナーゼファミリーは、メチル化シトシン(5mC)の5-ヒドロキシメチルシトシン(5hmC)への水酸化を触媒します。5hmCはDNMT1によってほとんど認識されないため、有糸分裂細胞で受動的DNA脱メチル化が起こるメカニズムを提供します(上記も参照)。ニューロンでは、5hmCはデアミナーゼによって認識され、5-ヒドロキシウラシル(5mhU)の形成を触媒します。この変化は、塩基除去修復(BER)機構によって修復されるU:Gミスマッチとして認識されます(図1.20-11)。BER複合体は、N-グリコシド結合を加水分解してアピリミジン/アプリン(AP)部位を作成するDNAグリコシラーゼで構成されており、APエンドヌクレアーゼによって除去され、結果として生じるギャップはDNAポリメラーゼによって埋められます。細胞増殖停止およびDNA損傷45タンパク質ファミリー(Gadd45a、Gadd45b、Gadd45c)は、デアミナーゼまたはグリコシル化酵素を5hmC部位にリクルートすることによってこのプロセスを促進します(図1.20-11)。興味深いことに、CpGジヌクレオチドにおけるCからTへの移行は、ヒト疾患で最も一般的な突然変異であり、この複雑なDNA脱メチル化/修復メカニズムが生物にとってかなりのコストと関連していることを示唆しています。この観察は、エピジェネティックコードと遺伝子コードの密接な関係をさらに強調しています。
クロマチンドメイン
クロマチンは伝統的にユークロマチン(活性クロマチン)とヘテロクロマチン(抑制クロマチン)に分けられてきました。しかし、最近のクロマチン沈降法とハイスループットシーケンシングの進歩により、より正確なクロマチンマッピングが可能になりました。この研究により、哺乳類細胞では、クロマチンがトポロジカルに会合するドメイン(TAD)に組織化されており、各染色体を約1Mbのセグメントに分割していることが示されています。TADは、活性クロマチン(ユークロマチンに相当)と、ポリコーム抑制クロマチン、ヌルクロマチン、構成的ヘテロクロマチンとして知られる3つの異なるサブタイプのヘテロクロマチンの4つの主要なカテゴリに分類できます(表1.20-5を参照)。異なるTAD間の境界は、活発に転写されているハウスキーピング遺伝子のCGIとインスレータータンパク質の結合部位によってかなり明確に区画されています。ただし、これらの境界内では、環境の合図に応じてある程度の柔軟性とシフトが発生する可能性があります。TADの位置と境界は遺伝子コードによってプログラムされており、したがって体内のすべての細胞で類似しています。しかし、各TAD内の正確なクロマチン内容は、異なる細胞型で異なります(図1.20-12)。TAD内容のこれらの変化は、ヒストン修飾、クロマチンリモデリング酵素、およびDNAメチル化によって誘発され、細胞が異なる環境条件に応じて遺伝子発現を変化させ、同じ遺伝子コードを使用して異なる細胞型を形成することを可能にします(図1.20-1)。このセグメント化された組織は、遺伝情報とエピジェネティック機構がどのように相互作用するかをさらに示しています。
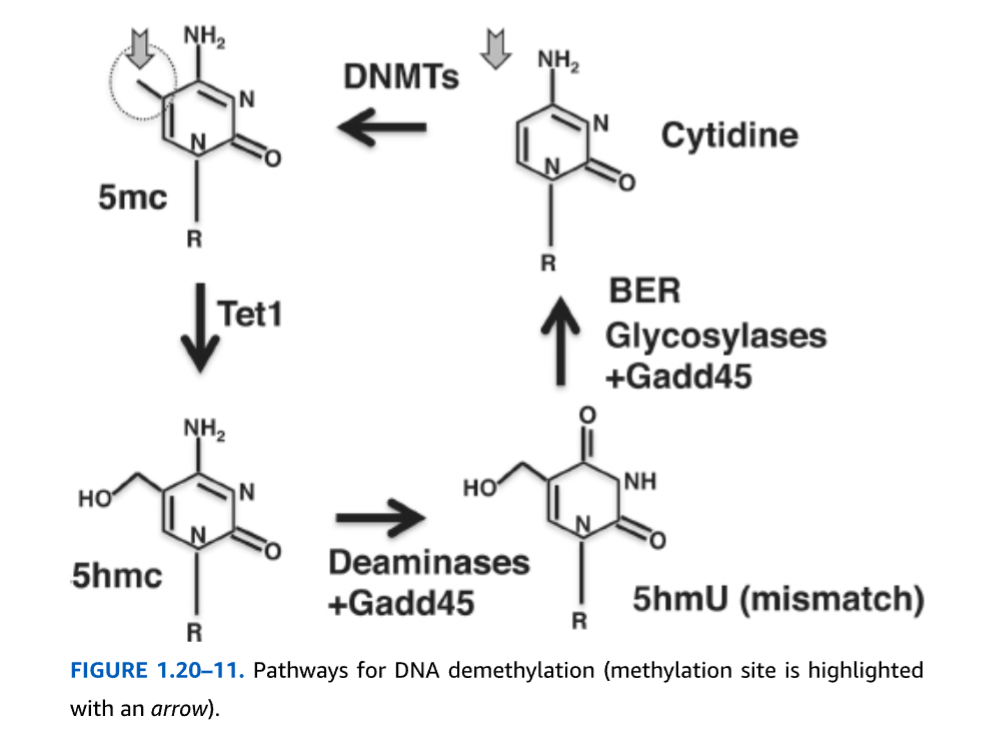
図 1.20-11. DNA脱メチル化経路(メチル化部位は矢印で強調されています)。
活性クロマチン
活性クロマチンは、オープンクロマチンと活性転写に関連する特定のヒストンアセチル化(H3K9-ac)およびメチル化(H3meK4)の存在によって特徴付けられます(表1.20-4参照)。活性TAD内の遺伝子は通常、共発現し、機能的に関連しています。活性TADは主に核の内部に存在し、場合によっては、効率的な「アセンブリライン」を形成するために、核の特定のサブ領域に集合します(図1.20-12)。おそらく最もよく特徴付けられている例は、核小体です。そこでは、リボソームRNA(rRNA)が合成され、リボソームタンパク質と結合してリボザイムを形成します。rRNA転写単位は、核小体形成領域(NOR)として知られる領域の異なる染色体上にクラスター化された、約100個のこのような単位のタンデムシリーズとして配置されています。すべてのNORが活性であるわけではなく、活性化プロセスは少なくとも部分的に、UBF転写因子がNORを核小体にリクルートする能力に依存しています。このリクルートプロセスは、rRNAクロマチンを活性状態に維持するのに役立ち、より効率的なリボソームのアセンブリを促進します。数百kbも離れたエンハンサー要素を、お互いに接触したり接触しなくなったりするようにクロマチンがループする能力、および転写機構は、クロマチンが環境の合図に応じて遺伝子発現を調節する重要なメカニズムを表しています(図1.20-12)。このようなループ形成がPTSD発症のリスクに与える影響については、臨床セクションで議論されています。
表 1.20-5.
クロマチンは4つの異なるトポロジカルに会合するドメインに分類できる
| TADクロマチン | ヒストン修飾および関連タンパク質 | 核/染色体上の位置 | クロマチン構造と転写活性化 |
| 活性 | H3K9-ac, H3meK4 | 核の内部 | オープンクロマチン、活性転写 |
| ポリコーム | H3K27-me3, PRC1, PRC2 | 分化した細胞の周辺 | コンパクトクロマチン、転写抑制 |
| ヌル | H3K9me2, 核ラミニン, H1ヒストン | 核膜 | コンパクトクロマチン、転写抑制 |
| 構成的/セントロメア周辺 | H4K20me3, H3K9me2/me3, HP1, H1ヒストン | クロモセンター、セントロメア、テロメア、タンデムリピート | 中程度の転写抑制を伴うコンパクトクロマチン |
図 1.20-12. 核内のクロマチンは、ハウスキーピング遺伝子またはインスレータータンパク質(図示せず)によって分離されたTADに組織化されています。TADは、活性、ポリコーム、ヌル、または構成的/ヘテロクロマチンに分類できます。各TADのクロマチン同一性は、異なる細胞型間または同じ細胞内の細胞周期全体で異なる場合があります。クロマチンの長距離ループは、遺伝子コード内で遠く離れたDNA領域をエピジェネティックコード内で近接させ、アセンブリライン(黒い点)の形成を可能にします。これらのクロマチンループは動的であり、環境の合図に応じて変化することができます(黒い矢印)。
ポリコーム抑制クロマチン
ポリコームTADは、**ポリコームグループ応答エレメント(PRE)**によって特徴付けられます。これらの配列は、PRC1およびPRC2複合体をこれらのTADにリクルートする特定の転写因子によって結合されます(図1.20-7)。したがって、ポリコームTADは、H3K27-me3、ポリコームタンパク質、および抑制されたクロマチンの存在によって特徴付けられます。ポリコームTADは、X染色体不活性化、インプリンティング、神経幹細胞分化、および皮質発達など、多くの重要な細胞プロセスを調節します。
ヌルクロマチン
哺乳類では、ヌルクロマチンはH3K9me2ヒストンメチル化によって特徴付けられます。この修飾は、TADヌルクロマチンをラミニンタンパク質を介して核膜にテザリングするために必要です(図1.20-12)。ヌルTADは、ヒトでは最大10Mbまで拡張する他のTADよりもかなり大きくなる可能性があります。これらは、高レベルのH1ヒストンとラミニンによって特徴付けられる、高度に抑制され、後期複製するクロマチンで構成されています。HDAC1はヌルクロマチンの一般的なサイレンシングに必要であるのに対し、HDAC3はヌルクロマチンを核膜にテザリングされた状態に維持する上で重要な役割を果たしているようです。ラミニンの変異は、ヌルクロマチンの核膜から核の中心部への移動を引き起こし、クロマチン脱凝縮、遺伝子活性化、およびハッチンソン・ギルフォード早老症候群などの重度の発達障害と関連しています。
構成的ヘテロクロマチン
構成的ヘテロクロマチンは、細胞周期全体で凝縮されており、通常、セントロメア、テロメア、およびタンデムリピートが豊富な領域の周辺に位置しています。ヒトクロマチンの約30%は構成的クロマチンであり、H4K20me3やH3K9me2/me3などのメチル化されたヒストン、特殊なヒストンバリアント、および高密度のHP1タンパク質の存在によって特徴付けられます(図1.20-6も参照)。構成的ヘテロクロマチンは、活性クロマチンよりも抑制的なクロマチンを提供しますが、ポリコームまたはヌルクロマチンと比較すると抑制性は低いです(図1.20-12)。
前臨床研究におけるエピジェネティクス
動物研究のいくつかの分野は、エピジェネティックな変化が、ストレス反応性、薬物探索行動、無力感行動など、神経回路および精神医学的に関連する表現型の安定した変化をどのように引き起こすかを見事に示しています。ここでは、エピジェネティックな変化が動物の複雑な行動をどのように修飾するか、およびこれらの発見の潜在的な治療への影響を説明するために、4つの例を議論します。
ストレス反応性と母性行動のエピジェネティック伝達
生命初期の経験が生体発達にとって重要であることは、精神力動的および生物学的の両方の観点から長らく認識されてきました。幼少期の虐待やネグレクトの事例は、しばしば世代間のパターンに当てはまり、子供時代に不適切に扱われた個人は、後の人生でしばしば適応不全の育児スタイルを示す傾向があります。初期の人生ストレス(ELS)への曝露は、成人期まで持続するストレスに対する内分泌および自律神経系の応答の亢進と関連しています。この亢進したストレス反応性は、感情障害、PTSD、薬物乱用を含むいくつかの精神病理を発症するリスク増加と密接に関連しています(精神疾患におけるエピジェネティックメカニズムを参照)。
ラットの「育児スタイル」の正常な異質性を利用した動物モデルは、親のネグレクトが成人期まで持続し、世代を超えて伝達されうる方法でストレス反応性を増加させる生物学的メカニズムを調査することを可能にしました。
生後最初の週のラットが子猫をなめたり毛づくろいしたりする頻度(LG)は、個体間で大きく異なり、一部の母親は他の母親のほぼ3倍の頻度で子猫をLGします。LGの頻度の違いは出産後の最初の週にしか見られないにもかかわらず、それらは子孫の多くの行動表現型に大きな影響を与えます(表1.20-6に要約)。例えば、高LGの母親によって育てられた子孫は、いくつかの記憶テストでより良い成績を収め、恐怖心が少なく、低LGの母親によって育てられた「ネグレクトされた」子孫と比較して、ストレスに対するコルチコステロン分泌の減少を示します。いくつかの観察は、これらの行動変化が、高LGと低LGの母親間の遺伝的差異ではなく、生後最初の週のLGの頻度によるものであることを示唆しています。第一に、交差飼育研究は、低LGの母親から生まれたが、高LGの母親によって育てられた子孫が、高LGの母親から生まれ育った子猫と行動的に区別できないことを示しています。同様に、高LGの母親から生まれたが、低LGの母親によって育てられた子孫は、低LGの母親の生物学的な子孫に似ています。第二に、生後最初の数週間にラットの子猫に異なる量の絵筆の刺激を与えることによって母性行動を模倣すると、同様の行動変化を誘発することができます。これらの明確な表現型は成人期まで持続し、生後最初の週に異なるレベルの触覚刺激に曝露することが、何らかの方法で成体の行動と生理をプログラムすることを示しています。重要なことに、高LGの母親によって育てられたメスの子孫は、それ自体が高LGの母親になり、低LGの母親から生まれたメスの子孫は低LGの母親になります(表1.20-6)。その結果、低LGと高LGの表現型は、生後早期の系統内で特定のレベルの触覚刺激を維持することによって、世代を超えて伝播します。
表 1.20-6.
高LGの母親と低LGの母親に曝露された子孫間の違い
| 成体期の行動表現型 | 高LG子孫 | 低LG子孫 |
| 視床下部-下垂体-副腎皮質軸 (HPA) 反応性 (GR抵抗性) | 低 | 高 |
| 不安 | 低 | 高 |
| 海馬依存性学習 | 健全 | 損なわれている |
| 母性行動 (LG) | 高 | 低 |
これらの観察研究は、生後わずか7日間の経験が、成体期のストレス反応性や母性行動をどのように決定するのかという疑問を提起します。その答えとして浮上しているのは、生後最初の週のLGの頻度が、ストレス反応性や母性行動を制御する神経回路における遺伝子の発現を調節するプロモーター要素のDNAメチル化に安定した変化を引き起こすというものです。例えば、グルココルチコイド受容体(GR)のプロモーターは、出生時には高度にメチル化されており、新生子ラットの海馬におけるGRレベルが低い原因となっています。生後最初の週に高LGに曝露されると、海馬においていくつかの一時的な変化が誘発され、最終的にGRプロモーターの広範な脱メチル化につながります。GRプロモーターの脱メチル化により、転写活性化因子である神経成長因子誘導性タンパク質A(NGFI-A)がGRプロモーターに結合できるようになります。NGFI-Aの結合は、HATとCBPをリクルートし、近くのヒストンをアセチル化させ、これがさらにクロマチンを開放し、高LG子孫の海馬におけるGR発現を増強します(図1.20-13)。高レベルのGRは、成体の高LG子孫の海馬に持続し、ストレスに応じてコルチコトロピン放出ホルモン(CRH)を分泌する視床下部室傍核(PVN)のニューロンに抑制的な影響を及ぼします。低LGの母親の子孫のGRプロモーターは過剰メチル化されたままです。この過剰メチル化は、NGFI-AがGRプロモーターに結合するのを妨げ、低LG子孫の海馬におけるGRレベルの低下につながります(図1.20-13)。要するに、生後最初の週の高LGへの曝露は、発達の重要な期間中にGRプロモーターを脱メチル化することにより、成人期のストレス反応性を低下させます。
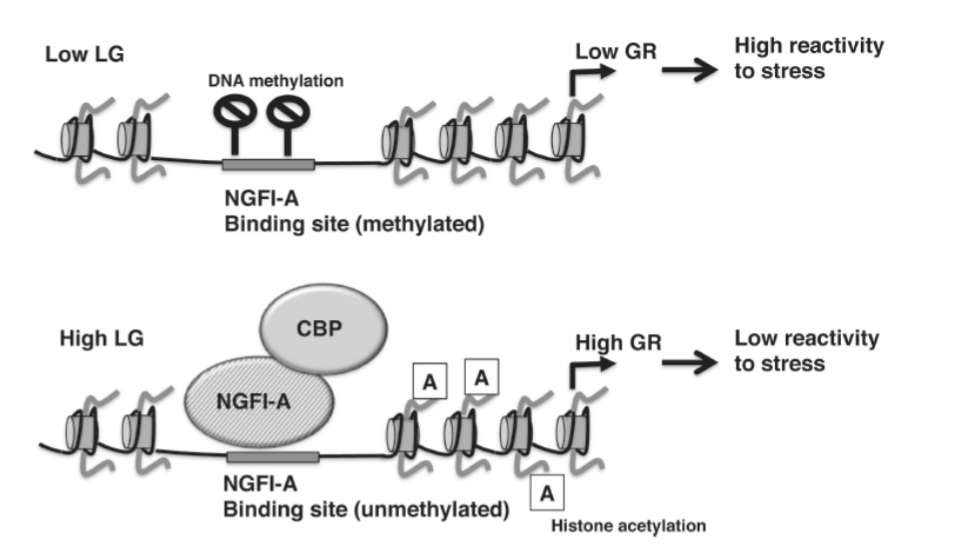
図 1.20-13. 上: 低LGの母親の子孫では、GRのプロモーターがメチル化状態に維持され、海馬でのGR発現レベルが低くなり、ストレス反応性が高くなります。下: 生後最初の週の高LGは、GRプロモーターの脱メチル化につながり、NGFI-A転写因子がHAT、CBPをリクルートして結合し、GR発現を活性化させます。海馬におけるGRレベルの上昇は、高LGの母親の子孫のストレス反応性を低下させます。
高LG行動への曝露は、視床下部の内側視索前野(MPOA)におけるエストロゲン受容体アルファ(Era)プロモーターのDNAメチル化も減少させます。このプロモーターの脱メチル化は、転写因子Stat5をEraプロモーターにリクルートし、高LGのメス子孫のMPOAにおけるEraレベルの増加を可能にします。低LGの母親によって育てられた子孫は、EraプロモーターにおけるDNAメチル化の持続により、MPOAにおけるEraレベルが低いままです。MPOAにおけるEraレベルの違いは成人期まで持続し、オキシトシン発現を調節し、成体のメス子孫におけるその後のLG行動をプログラムすると考えられています。
LGがDNAメチル化、遺伝子発現、および行動表現型に安定した変化を引き起こす能力に関する重要な臨床上の疑問は、これらの変化が成人期に薬理学的に元に戻せるかどうかです。この質問に対する答えは「はい」のようです。例えば、HDAC阻害剤の脳室内注入は、GRプロモーターのDNAメチル化を減少させ、海馬のGRレベルを増加させ、成体の低LG子孫のストレス反応性を減少させます。HDAC阻害剤はDNAメチル化を直接変化させませんが、クロマチンアクセシビリティを増加させ、このプロセスはDNAの脱メチル化を促進する可能性があります。逆に、メチオニン(メチル化反応のドナー)の脳室内注入は、GRプロモーターのDNAメチル化を増加させ、海馬のGR発現を減少させ、成体の高LG子孫のストレス反応性を増加させます。これらの知見は、GRプロモーターのエピジェネティックランドスケープが、オープンクロマチンを促進する活動(ヒストンアセチル化、DNA脱メチル化)とクロマチンをコンパクト化する活動(メチル化剤)のバランスを傾けることによって、成人期に薬理学的に修飾できることを示唆しています。HDAC阻害剤またはメチオニンの長期間のウォッシュアウト後に、GRプロモーターにおけるLGによって誘発された発達表現型が再確立されるかどうかは、現在不明です。この問題に取り組むことは、これらの発達的に敏感なエピジェネティックな変化の真の可逆性に関して重要な意味を持つでしょう。
これらの発見のヒトの精神病理学への関連性は、小児期の虐待やネグレクトの既往歴がある自殺犠牲者の脳と、小児期のトラウマがない自殺犠牲者の脳を比較する死後研究によって検証されてきました。ラットにおける前臨床研究と同様に、小児期の不適切な扱いは、GRプロモーターにおけるDNAメチル化の増加と海馬におけるGRレベルの低下と関連していました。生きた患者におけるエピジェネティックシグネチャを評価する課題は依然として残っていますが(後述の「精神疾患におけるエピジェネティックメカニズム」のセクションを参照)、これらの研究は、げっ歯類における前臨床研究が、エピジェネティクスがヒトの精神状態にどのように寄与するかのメカニズムに関する重要な洞察を提供できることを示唆しています。
マウスにおけるプラダー・ウィリー症候群の救済
プラダー・ウィリー症候群(PWS)は、約15,000人から30,000人に1人の割合で影響を及ぼすインプリンティング障害です。この多様な症候群の主要な特徴には、新生児期の筋緊張低下と発育不全があり、その後、過食症、小児肥満、性腺機能低下症、軽度の知的障害が続きます(表1.20-3)。PWSは、父親由来の15番染色体(例:15q11-q13)に位置する一群の遺伝子の発現欠如によって引き起こされます。この領域には、ネクジン(Necdin:NDN)、小型核リボヌクレオタンパク質ポリペプチドN(SNPRN)、ユビキチンプロテインリガーゼE3A(UBE3A)などのいくつかのタンパク質コード遺伝子、およびSNORD116やUBE3A-ATSなどの非コードRNAが含まれており、これらの発現はPWSインプリンティングセンター(PWS-IC)によって調節されています。男性では、PWS-ICのDNAは非メチル化されており、オープンクロマチン状態に維持されているため、PWS遺伝子が発現できます(図1.20-14、上)。男性におけるUBE3A-ATSの発現は、その隣接するUBE3A遺伝子の局所的な抑制を引き起こします(図1.20-14、上)。母親染色体では、PWS-ICのDNAは高度にメチル化されており、ヒストンメチラーゼG9Aのリクルートにつながります。G9Aはヒストン3のリジン9(H3K9-Me2およびH3K9-Me3)をメチル化し、結果としてPWS遺伝子座全体にコンパクトなクロマチンが拡大し、母親染色体からの遺伝子発現が遮断されます(図1.20-2、中)。母親染色体上でのUBE3A-ATS発現の欠如は、UBE3A遺伝子の転写を可能にします。これは重要です。なぜなら、母親染色体からの機能的なUBE3Aの発現を損なう状態は、アンジェルマン症候群を引き起こすからです(表1.20-3)。
父親由来の15q11-q13遺伝子座の欠失は、これらの遺伝子の唯一のゲノム源を排除し、PWSの最も一般的な原因です。これらの各遺伝子がPWSにどの程度寄与しているかはまだ解明されていませんが、非コードで高度に保存されたRNAであるSNORD116が病理に重要な寄与をしているという研究が増えています。母親染色体上に完全ではあるが高度にコンパクト化されたクロマチンが存在するという観察は、母親クロマチンを開放させることでPWSの臨床的特徴を排除するか、少なくとも軽減できる可能性を示唆しました。これを達成するために、母親由来のPWS-ICから緑色蛍光タンパク質(GFP)を発現するトランスジェニックマウスから胚性線維芽細胞が採取されました。母親由来のPWS-ICは不活性であるため(図1.20-14、中)、この細胞株ではGFPの発現は見られません。しかし、9000以上の小分子からなる大規模なライブラリに曝露したところ、2つの化合物(UNC0638とUNC0642)がこの細胞株でGFPの発現を誘発することができました。両化合物は既知のHMT G9A阻害剤であり、ヒストンメチル化の遮断が母親染色体上のコンパクトなクロマチン構造を開放する鍵となる可能性を示唆しています。さらなる分析により、UNC0638とUNC0642が母親遺伝子座のクロマチン構造を開放し、マウス胚性線維芽細胞におけるPWS遺伝子の発現を回復させることが確認されました。PWS患者から得られた線維芽細胞をUNC0638とUNC0642に曝露した場合にも同様の結果が得られました。このアプローチの特に有望な点は、DNAメチル化を遮断する薬剤(5-アザ-dcなど)とは異なり、UNC0638とUNC0642がSNORD116を含むPWS遺伝子座に見られるほとんどの遺伝子の発現を増加させたことです。さらに、PWSのマウスモデルの治療では毒性の証拠はなく、このモデルにおける発育不全による死亡率が著しく減少しました。UNC0638とUNC0642によるin vivoおよびin vitroでの治療は、UBE3Aの発現を遮断しアンジェルマン症候群を引き起こすような、より下流の遺伝子であるUBE3A-ATSの発現を活性化しませんでした(図1.20-14、下)。UNC0638とUNC0642はヒストンメチル化を遮断しましたが、DNAメチル化を減少させたり、母親PWS-IC領域でのG9A結合を妨げたりしませんでした。これは、ヒストンメチル化の遮断が、高レベルのDNAメチル化が存在する場合でも、一部の遺伝子発現を回復させ、致死性を低下させることができることを示唆しています。母親染色体からの遺伝子発現は、父親遺伝子座から発現されるレベルの30-50%にしか達しませんでしたが、これはDNAメチル化とヒストンメチル化の両方が完全な抑制に必要であり、このシステム内の1つの調節層を逆転させるだけではPWSの部分的な救済にしかならない可能性を示しています。それでも、レポーター媒介細胞株と小分子のハイスループットスクリーニングを組み合わせて局所的なクロマチン構造の変化を誘発するこの方法は、エピジェネティック創薬におけるエキサイティングな新しいフロンティアを提供します。
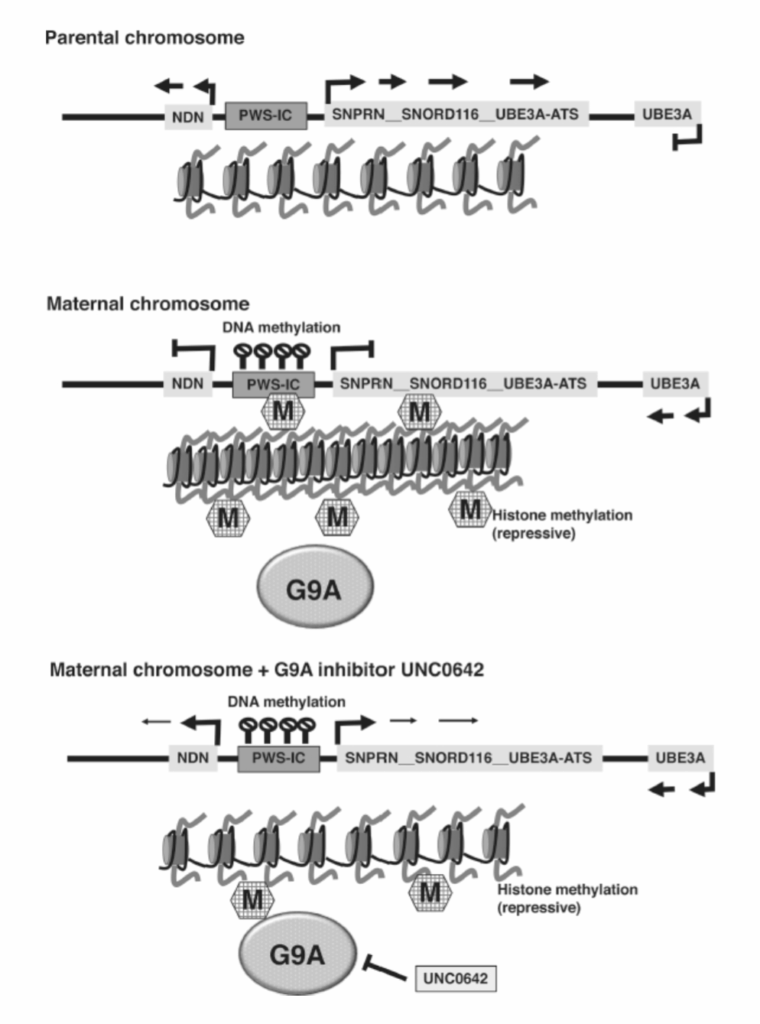
図 1.20-14. 上: プラダー・ウィリス・インプリンティング・センター(PWS-IC、濃い灰色で示される)の父親由来DNAはメチル化されておらず、オープンクロマチンの形成と、正常な神経発達に必要なNDN、SNPRN、SNORD116などの遺伝子の発現を可能にします。UBE3A-ATSの発現は、父親由来染色体からのUBE3A転写を妨げます。中: PWS-ICの母親由来DNAはメチル化されており、HMT G9Aをリクルートし、PWS遺伝子座全体にヒストンメチル化とクロマチンコンパクト化をもたらします。コンパクト化されたクロマチンは、UBE3-ATSを含む遺伝子発現を妨げますが、母親染色体からのUBE3Aの転写は回復させます。下: UNC0642はG9A HMT活性を阻害し、母親PWS遺伝子座からのNDN、SNPRN、SNORD116などの遺伝子の部分的な発現を可能にし(ただしUBE3-ATSは含まない)、PWSのマウスモデルにおける仔の死亡率を減少させます。
うつ病行動と抗うつ薬作用のエピジェネティックメカニズム
うつ病とPTSDのげっ歯類モデルは、典型的には、絶望、回避、または快感欠乏様行動につながる、慢性的、予測不可能、または重度のストレスの何らかの形態を利用します。そのようなパラダイムの1つは、マウスを10日間にわたって攻撃的な侵入者に短時間曝露する慢性社会的敗北を含みます。敗北したマウスは、ショ糖摂取量の減少を示し、快感欠乏様状態を示唆し、強制水泳および尾懸垂試験において無力感が増加します。ヒトのうつ病と同様に、これらの行動変化の多くは、慢性的な抗うつ薬治療によって逆転しますが、急性治療では逆転しません。
いくつかの観察は、社会的敗北の行動的結果と、抗うつ薬によるうつ病表現型の回復が、神経可塑性に関与する遺伝子の発現におけるエピジェネティックな変化によるものであることを示唆しています。神経可塑性遺伝子のエピジェネティック調節の最も良い例の1つは、辺縁系脳領域における脳由来神経栄養因子(BDNF)の発現に対するストレスと抗うつ薬の相反する効果です。海馬におけるBDNF発現は、敗北したマウスでダウンレギュレートされ、ストレス終了後何週間も持続します。この効果は、三環系抗うつ薬イミプラミンによる慢性治療で逆転させることができます。社会的敗北が海馬BDNFレベルを低下させる能力は、BDNFプロモーターにおけるH3K27メチル化の増加によって媒介されるようです。これらのヒストン修飾は、BDNF発現を阻害する抑制的なクロマチン状態につながります(図1.20-15参照)。これらの知見は、うつ病患者がBDNFプロモーターにおいてH3K27ジメチル化(H3K27-me2)の増加とBDNFレベルの低下を示すというヒトの死後研究と一致しています。イミプラミン治療は、敗北したマウスの海馬におけるうつ病表現型を回復させ、BDNFレベルを増加させましたが、H3K27-me2レベルは変化させませんでした。代わりに、イミプラミン治療は、BDNFプロモーターにおけるH3K9残基のアセチル化を増加させました。これは、オープンクロマチンと転写活性化に関連する修飾です(図1.20-15)。これらの結果は、アセチル化がH3K27-me2の抑制的な性質を上書きできる、ヒストンコードの組み合わせ的な性質を強調しています。イミプラミン治療は、対照マウスではなく敗北したマウスでHDAC5の発現を減少させ、敗北したマウスにおけるHDAC5の減少がH3K9アセチル化を促進し、したがってイミプラミンの治療効果を媒介する可能性を提起しました(図1.20-15)。この主張は、海馬におけるHDAC5の過剰発現が、敗北した動物におけるイミプラミンの抗うつ効果をブロックすることを示すデータによって裏付けられています。これらの前臨床的知見は、HDAC阻害剤による治療が、BDNFを増加させ、うつ病を治療するための新しい治療戦略を提供する可能性があることを示唆しています。しかし、多大な努力にもかかわらず、現在そのような治療法は利用可能ではなく、精神状態を治療するためにエピジェネティック修飾剤を使用することの複雑さと課題を浮き彫りにしています(この問題については以下で詳述)。
コカイン依存症のエピジェネティックメカニズム
これまでのセクションでは、負の人生経験(恐怖条件付け、慢性社会的敗北、または母性のネグレクト)がクロマチン構造に長期的な変化を引き起こす方法について議論しましたが、強烈な報酬経験もエピジェネティックにコード化される可能性があります。典型的な例は薬物依存症であり、そこでは自然な報酬システムが非常に報酬的な外来物質によって乗っ取られ、適応不全で強迫的な薬物探索行動を引き起こします。
報酬経験によって駆動される薬物摂取の初期実験段階から、強迫的で適応不全な行動によって特徴付けられる依存症段階への移行は、ヒトからげっ歯類まで多様な種で見られる漸進的なプロセスです。げっ歯類では、依存症行動への移行は、報酬システム内に位置するニューロンにおけるシナプス可塑性を修飾する遺伝子の発現におけるエピジェネティックな変化によって媒介されます。このセクションでは、このプロセスについて学んだ主要な教訓の一部を簡潔にまとめ、側坐核(NAc)におけるヒストンアセチル化が薬物探索行動の発達に果たす役割に焦点を当てます。
図 1.20-15. 上: 対照マウスは正常なBDNFレベルと多幸様表現型を示します。中: 繰り返される社会的敗北への曝露は、抑制的なHK27-me2メチル化(窓枠模様の六角形)の増加につながり、クロマチン凝縮、BDNFレベルの低下、うつ病様表現型を引き起こします。下: 社会的に敗北したマウスのイミプラミンによる治療は、HK27-me2レベルを変化させませんが、HDAC5レベルを減少させます。HDAC5レベルの低下は、ヒストンアセチル化(灰色の四角)の増加、クロマチンの脱凝縮、BDNFレベルの増加、および多幸表現型の回復に寄与します。
コカインの即座の報酬効果は、いくつかの脳回路におけるドーパミンレベルの増加と密接に関連しており、側坐核(NAc)はその中でも最も重要でよく研究されている標的の1つです。コカインの初期の報酬効果は数分以内に経験され、c-fosなどの即時早期遺伝子の一時的な発現につながります。慢性的だが急性ではないコカインの投与は、FosB/△FosB、CDK5、BDNFなどの追加の遅発性遺伝子群の発現を増加させるために必要です。これらの遅発性遺伝子の多くは、コカインが存在しなくても何週間も発現し続けることができ、げっ歯類において強力な薬物探索行動を引き起こすために必要です。この効果は、非常に安定した転写因子である△FosBについて広範に研究されてきました。c-fosの繰り返し活性化は、NAcニューロンのサブセットにおける△FosBの蓄積につながります。高レベルの△FosBは、NAcニューロンにおけるシナプス可塑性を増強するシナプスタンパク質を発現させるために必要であり、これが薬物探索行動に必要な安定した変化をもたらします。△FosBの蓄積は、運動や摂食などの他の強迫的な摂取行動でも見られ、コカインが内因性の報酬経路を乗っ取ることができるという考えと一致しています。
ヒストンアセチル化は、即時早期遺伝子の誘導から△FosBなどの遅発性遺伝子の安定した発現への移行を調整する上で重要な役割を果たします。急性コカイン投与は、H4アセチル化とH3リン酸化を増加させることによってc-fosの発現を誘導します。コカインを繰り返し使用すると、これらのクロマチン変化はc-fosプロモーターではもはや存在せず、c-fosレベルは低下します。代わりに、繰り返しのコカイン投与は、遅発性遺伝子(例:FosB、△FosB、Cdk5、BDNF)のプロモーターにおけるH3アセチル化の増加につながります。HAT CBPのウイルスノックダウンやNAcにおける異なるHDACの過剰発現など、ヒストンアセチル化を阻害する操作は、慢性コカインが遅発性遺伝子を誘導する能力をブロックし、強迫的な薬物探索行動を減少させます。これらの知見は、薬物依存症への移行が、最初の急速なH4アセチル化から、遅発性遺伝子を誘導するために必要なより遅いH3アセチル化の波へのエピジェネティックなスイッチによって媒介されるというモデルを示唆しています。
ただし、これは非常に単純化されたモデルであり、慢性コカインのエピジェネティックな影響ははるかに複雑であることに注意することが重要です。例えば、NAcにおけるHDAC1の長期的な阻害は、コカインの運動活動への感作を減少させますが、これはヒストンアセチル化の「予想される」増加と薬物探索行動の促進とは矛盾します。この予期せぬ結果は、慢性的なHDAC1阻害が、慢性コカイン曝露後でもGABA受容体遺伝子のプロモーターにおける抑制的なH3K9-me2マーカーを維持する能力によるものと考えられます。したがって、H3K9-me2抑制マーカーの保持は、高レベルのアセチル化が存在する場合でもこれらの遺伝子の発現を妨げます。この例は、異なるヒストン修飾がクロマチン構造と遺伝子発現を修飾するためにどのように相互作用するかの複雑さを強調しています(上記の図1.20-15も参照)。
慢性コカインはまた、NAcにおけるDNMT3aの発現をダウンレギュレートし、DNMT3aを減少させる操作は依存症表現型を増強します。これらの知見は、コカイン依存症の行動的結果の一部が、NAcにおけるDNAの脱メチル化によって媒介されることを示唆しています。最後に、コカイン投与後に見られるエピジェネティックな変化の一部は、細胞型特異的であるようです。例えば、D1ニューロンにおけるHMT(G9A)のノックアウトは、コカインの行動的結果を減少させますが、同じ酵素がD2ニューロンでノックアウトされた場合には、逆の表現型が見られます。異なる細胞型が非常に異なるクロマチン構造と機能を持っていることを考えると(導入と図1.20-1を参照)、この細胞型特異性は驚くべきことではありません。しかし、この細胞型特異性は、特定のクロマチン活性(例:HMT G9A)を標的とする薬理学的介入や、NAcのような不均一な脳領域で見られるクロマチン変化を解釈する上での課題を表しています。
精神疾患におけるエピジェネティックメカニズム
精神医学研究におけるエピジェネティクスは、精神医学研究におけるいくつかの基本的な未解決の疑問に対処する上で大きな期待を抱かせますが(表1.20-2)、近年、その臨床実装において重大な課題も浮上しています。この最終セクションでは、精神医学的ケアを革新するためのエピジェネティック研究が持つ可能性と課題を説明する3つの臨床例を提供します。
脆弱X症候群のエピジェネティック病因
脆弱X症候群(FXS)は、世界の遺伝性知的障害の主要な原因であり、男性の推定有病率は1:2,500です。これは、中程度の知的障害、社会的欠陥、ADHDの発生率の高さ、および複数の身体的異常を特徴とするX連鎖単遺伝性神経発達精神障害です(表1.20-3)。脆弱X症候群は、脆弱X精神遅滞遺伝子1(FMR1)の機能喪失によって引き起こされます。FMR1遺伝子は、正常なシナプス発達と認知に不可欠なシナプスおよびクロマチン修飾遺伝子のレベルを調節するRNA結合タンパク質をコードしています。「単遺伝性」という用語は突然変異した遺伝子コードを意味しますが、実際の疾患原因メカニズムは、この遺伝子のプロモーターをサイレンシングするエピジェネティックな変化を伴います。
FXSにおけるCGGリピートの数は、正常、前突然変異、完全突然変異アレルの3つのグループに分類できます。正常アレルは5〜55個のCGGリピートからなり、遺伝子の正常な発現を可能にします。前突然変異アレルは56〜200個のリピートを持ち、FMR1タンパク質の正常な発現を維持します。しかし、前突然変異アレルは、さらなる伸長のリスク増加と関連しており、200個以上のリピートと定義される完全突然変異領域に押し込まれる可能性があります。各CGGリピートはCpGジヌクレオチドを含んでいるため、この伸長したリピートは不安定なゲノム領域として認識され、高度にメチル化されます。他の高度にメチル化されたCpGアイランドの場合と同様に(エピジェネティックな機構、DNAメチル化のセクションを参照)、このメチル化された領域は、FMR1遺伝子の発現を妨げる高度に抑制的なクロマチン構造を形成する追加のリモデリング酵素をリクルートします(図1.20-16)。
まれなケースのサブセットでは、完全突然変異アレル(すなわち、200以上のリピート)を持つ個人でもFXSの臨床症状を発症しません。これらのまれなケースでは、伸長したリピートが何らかの形でDNAメチル化を逃れており、完全な突然変異を持つ個人において臨床症候群を誘発するためにDNAメチル化が必要であるという説得力のある証拠を提供しています。したがって、DNAメチル化を予防する治療法は、完全突然変異アレルを持つ個人の神経発達上の後遺症を潜在的に救済できる可能性があります。実際、FXSを持つ個人から得られた細胞株およびiPSCを、5-アザシチジンなどのDNA脱メチル化剤で治療したところ、FMR1タンパク質を部分的に脱メチル化し、発現させることができました。しかし、これらの化合物の臨床的有用性は、その高い細胞毒性と突然変異のリスクのために限られており、無毒で安定した、高度に特異的なエピジェネティック介入の開発が必要です。実際、最近の概念実証研究では、FXS患者由来のiPSCからCGGリピートを精密に切り出すためにクリスパー技術が成功裏に使用され、FMR1遺伝子の完全な活性化と、これらのiPSCから生成されたニューロンにおける正常な電気生理学的特性の回復につながりました。臨床的有用性の観点からははるかに将来の話ですが、この研究は、ヒトにおける新しいエピジェネティック介入の結果をテストする上でのiPSCの有用性を示しています。
遺伝子と環境の相互作用がFKBP5の発現とPTSDのリスクを調節する
多くの場合、ストレスが複雑な行動に与える影響は、遺伝的脆弱性によって媒介されます。この種の遺伝子と環境の相互作用のいくつかの例が精神医学文献に記述されています。しかし、これらの相互作用が起こる分子の詳細については、まだ明らかになり始めたばかりです。これは最近の一連の研究で示されており、小児期のトラウマが特定の遺伝的素因と優先的に相互作用して、FK506結合タンパク質5(FKBP5)の発現とPTSDおよびうつ病への脆弱性を増加させる方法が示されています。
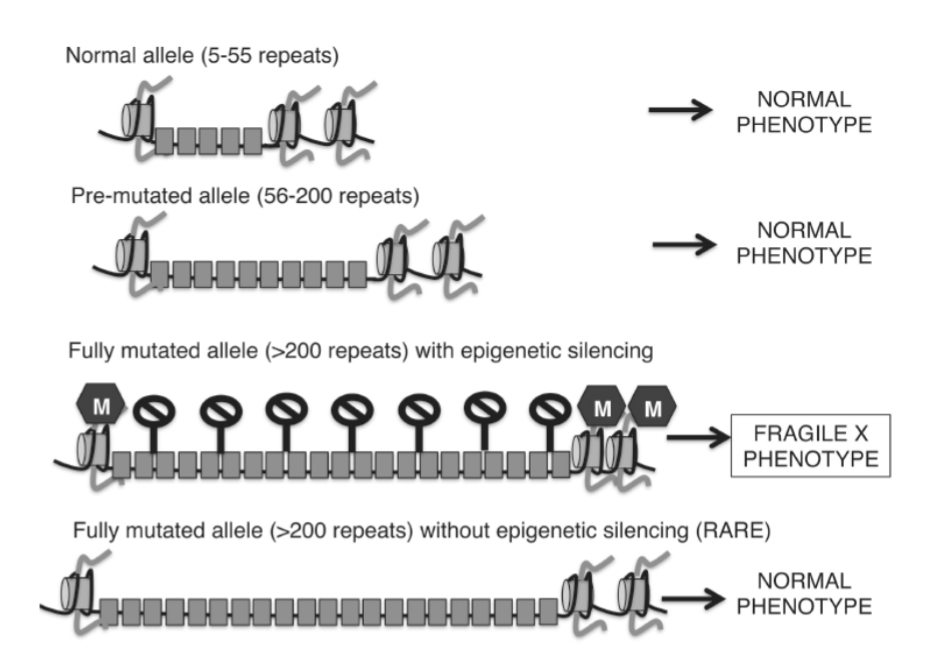
図 1.20-16. 正常な個人では、FMR1遺伝子には5〜55個のCGGリピートが含まれています(各灰色の箱は10個のCGGリピートを表します)。前突然変異を持つ個人は56〜200個のリピートを持ち、表現型は正常です。しかし、彼らは減数分裂中にリピート伸長のリスクが高く、その結果、子孫に完全な突然変異表現型が生じます。完全突然変異を持つ個人(200個を超えるCGGリピート)のDNAプロモーターは高度にメチル化され(黒い進入禁止標識で示される)、FMR1発現が異常に減少し、脆弱X表現型をもたらします。まれに、完全な突然変異を持つにもかかわらずエピジェネティックサイレンシングを欠いている個人が特定されています。これらの子供は表現型が正常であり、DNAメチル化が臨床症候群を誘発するために必要であることを示唆しています。
FKBP5は、GRとの相互作用を介してその効果を発揮するストレス関連遺伝子です。基礎条件下では、GRは熱ショックタンパク質90(Hsp90)とFKBP5タンパク質によって細胞質に維持されており、これらはGRに結合し、その核局在化シグナルを隠します。ストレスによって誘発される高レベルのコルチゾールは、Hsp90とFKBP5をGRから解離させ、GRが核に入り、2つの転写プログラムを誘導することを可能にします。最初の転写プログラムは、副腎からのコルチゾールのさらなる放出を停止させるために不可欠です(ストレス反応性と母性行動のエピジェネティック伝達のセクションを参照)。2番目のタスクは、GRの核へのさらなる進入を防ぐ負のフィードバックループであるFKBP5の誘導を含みます。GRを細胞質に保持することは、ストレス応答をリセットし、追加の脅威に反応して再活性化できるようにすると考えられています(図1.20-17)。
FKBP5が過剰発現する状況では、GRの正常な活性化が妨げられ、ストレス応答の非効率な停止につながります。HPA活性を停止できないこの状態は、高レベルのコルチゾールへの長期曝露につながり、「GR抵抗性」と呼ばれます。GR抵抗性は、うつ病、PTSD、および他のいくつかの医学的合併症のリスク増加と関連しています(図1.20-17)。この疾患モデルは、インスリン抵抗性と同様です。インスリン抵抗性では、インスリンシグナル伝達の非効率性が、循環中の高レベルのグルコースを維持することによって末梢臓器毒性を引き起こします。
FKBP5遺伝子のrs1360780ハプロタイプには、A/TアレルとC/Gアレルがあります。ベースラインでは、A/TアレルはGR誘導FKBP5発現のレベルが高く、保護的なC/Gアレルと比較してより堅牢なHPA活性化につながります(図1.20-18)。A/Tアレル単独ではストレス関連精神障害のリスク増加とは関連していませんが、このリスクアレルは幼少期のトラウマと用量依存的に相互作用し、PTSDのリスクを増加させます。言い換えれば、A/Tアレルを持ち、より重度の小児期の不適切な扱いに曝露された個人は、PTSDとうつ病を発症する可能性が高いです。これは、小児期のトラウマへの曝露がPTSDやうつ病のリスク増加と関連していない保護的なC/Gアレルを持つ個人には当てはまりません。
これらの臨床観察のメカニズム的説明は、FKBP5レベルとGR抵抗性の遺伝的およびエピジェネティックな調節間の相乗効果を示唆しています。遺伝子レベルでは、A/T感受性を持つ個人は、より効率的なGR媒介FKBP5を示し、C/G個人よりも高レベルのFKBP5を産生します。FKBP5遺伝子には、ストレスに応じてGRが占有する2つの**グルココルチコイド応答エレメント(GRE)**があります。1つはイントロン2(GRE-2)にあり、転写開始部位(TSS)から約50kb離れています。もう1つのGREはイントロン7(GRE-7)にあり、FKBP5のTSSから約100kb離れています。これらの長い距離にもかかわらず、両方のGREは長距離ループを介してTSSと接触することができます。これらのクロマチン媒介ループは、これらのGREをTSSの近くにリクルートし、FKBP5発現を駆動します。GRが活性化されると、GRE-7部位は遺伝子型に関係なくTSSにリクルートされます。対照的に、GRE-2はAT個人でのみTSSにリクルートされます。GRE-2がTSSの近くに存在することは、ストレスに対するFKBP5のより堅牢な発現、したがってA/Tアレルを持つ個人におけるより大きなHPA活性化につながります(図1.20-18)。これらの遺伝子変化がこれらの異なるループ構成にどのように寄与するかは現在不明ですが、これらのループの存在はクロマチンの重要な機能です(活性クロマチンのセクションを参照)。
これらの遺伝的差異の結果として、幼少期の虐待に曝露されたA/T個人は、C/G個人と比較して高レベルのコルチゾールを分泌すると予測されます。コルチゾールレベルの増加は、リスクアレルを持つ個人においてGRE-7結合部位のDNAメチル化を減少させると考えられています。GRE-7のDNAメチル化の減少は、GRのより効率的なリクルートにつながり、幼少期のトラウマに曝露されたA/T個人におけるFKBP5発現とGR抵抗性をさらに増強します(図1.20-18)。GRE-7は、虐待またはネグレクトを受けたG/C個人ではメチル化されたままであり、このエピジェネティックマーカーの特定の遺伝子-環境相互作用を示しています。患者由来の単球を用いたex vivo実験では、GRE-7メチル化とグルココルチコイド効力の間に強い負の相関が示されており、このエピジェネティックバイオマーカーがGR抵抗性を媒介する機能的役割を支持しています。
トラウマがこれらのエピジェネティックな変化を誘発する能力は、敏感な期間に限定されているようです。成人期のトラウマへの曝露は、GRE-7メチル化率の減少にはつながりません。これらの臨床的知見は、増殖中のNSCではデキサメタゾンがGRE-7メチル化を減少させるが、完全に分化した分裂後のニューロンでは減少させないという細胞培養実験によって裏付けられています。増殖中に曝露された場合、このメチル化の減少は、NSCがニューロンに分化した後も、デキサメタゾン刺激が除去された後でも維持され、脱メチル化されたGRE-7が分化したニューロンに安定して伝播することを示唆しています。これらの観察は、高レベルの神経新生が存在する小児期のストレス曝露が、この遺伝子と幼少期ストレスの相互作用の原因であることを示唆しています。したがって、成人期のストレスを除去する介入と比較して、幼少期のストレスを軽減することを目的とした介入の方が、PTSDへの脆弱性を軽減する上でより効果的である可能性が高いです。
図 1.20-17. A: ストレスがない場合、コルチゾールレベル(「C」の三角形)が低いとき、グルココルチコイド受容体(GR)はシャペロンタンパク質FKBP5とHSP90に結合しています。この結合状態では、GRは核に入り、転写を活性化するのを妨げられます。B: ストレスに曝露されると、コルチゾールレベルが増加し、FKBP5/HSP90複合体を置き換えることでGRが核に入ることが可能になります。核内で、GRはさらなるコルチゾール放出を停止させる遺伝子の転写を活性化し、FKBP5の転写を活性化します。FKBP5レベルの増加は、GRのさらなる核への進入を防ぎ、システムをリセットして次のストレスの波に対応できるようにします。C: FKBP5の過剰発現は、高レベルのコルチゾールがGRを核に駆動するのを妨げることでGR抵抗性をもたらします。その結果、追加のコルチゾールが放出され、循環中のコルチゾールレベルが高くなります。
エピジェネティックな自殺シグネチャー:バイオマーカーへの前進
自殺は、米国における死因の第11位です。死亡前に自殺者の約半数が精神保健従事者によって診察されているという観察は、正確なリスク評価の重要性を浮き彫りにしています。自殺を予測する信頼できるバイオマーカーがないため、精神医学的評価は、致命的な自傷行為の可能性を評価するために、長いリストの危険因子に依存しています。これらの集団リスク因子は予測値が低く、これらのリスクを持つほとんどの人は自殺せず、最終的に自死する一部の個人はこれらのリスクを共有しない場合があります。したがって、特定の末梢エピジェネティックマーカーが80%以上の精度で自殺を予測できるという最近の発見は、これらの悲劇を防ぐための画期的な可能性を提供します。
自殺のエピジェネティックマーカーを探索するために、自殺したうつ病患者の前頭前皮質から得られたDNAメチル化パターンが、他の原因で死亡したうつ病患者と比較されました。DNAメチル化は、その相対的な安定性と遺伝子発現を予測する能力のため、死後組織での研究に適しています(DNAメチル化を参照)。紡錘体および動原体関連複合体サブユニット2(SKA2)をコードする1つの遺伝子のみが、対照群と比較して自殺遂行者のニューロンで差次的にメチル化されていることが判明しました。SKA2は、GRを核にシャペロンし、したがってストレスに対するHPA活性化を停止させるのに役立つ足場タンパク質です。SKA2の3’非翻訳領域(3′-UTR)におけるDNAメチル化の増加は、別の死後コホートで再現され、信頼できる自殺の遡及的バイオマーカーであることを示唆しています。SKA2のメチル化は、SKA2タンパク質レベルの減少と関連しており、このエピジェネティックな修飾が自殺者におけるSKA2活性を調節していることを示唆しています。興味深いことに、SKA2の3′-UTRにあるCpGメチル化部位には、シチジンがチミジンに置き換わった代替の一塩基多型アレルが含まれています。このTアレルはメチル化されず、したがって保護アレルと考えられています。
死後での再現は重要な第一歩ですが、より重要な問題は、末梢組織におけるこのエピジェネティックバイオマーカーの存在が、生きている患者における将来の自殺行動を予測できるかどうかです。これは、まず、死後分析から得られた前頭前皮質ニューロンおよびグリア細胞における遺伝子発現と、うつ病患者の異なるコホートから得られた末梢免疫細胞における遺伝子発現を比較することによって評価されました。この分析は、ニューロンにおける遺伝子発現と末梢免疫細胞との間に高い相関を示しましたが、グリア細胞とは相関せず、2つの細胞集団が類似のエピジェネティックシグネチャを共有していることを示唆しており、これは末梢バイオマーカーを確立する上で重要な特徴です。次に、末梢免疫細胞におけるSKA2メチル化状態、遺伝子構成(T対Cアレル)、および自殺念慮の関係が、3つの異なるコホートで前向きに評価されました。注目すべき発見は、自殺念慮の予測において、遺伝的素因(T対Cアレル)とエピジェネティック状態(低メチル化対高メチル化)との間に有意な相互作用があったことです。さらに、免疫細胞におけるSKA2メチル化レベルは、朝の(覚醒時の)コルチゾールレベルと強く相関しており、SKA2メチル化とストレスレベルとの間の可能な機能的関連性を示唆しています。場合によっては、SKA2における高レベルのメチル化が、自殺念慮の報告された発症の数ヶ月前に検出され、このバイオマーカーが自殺念慮を発症する前にこれらの個人に存在していた可能性を提起しています。
コルチゾールレベルや不安などの追加変数を組み込むことで、モデルの予測値がさらに向上しました。この改訂されたモデルは、遺伝子型(T対Cアレル)、SKA2のメチル化率、および不安スコアを使用して、別の前向きコホートでテストされました。このモデルは自殺行動を71%の精度で予測でき、唾液コルチゾールレベルが追加された場合には82%に増加しました。自殺念慮の分類の厳密さをさらに改善すると、予測値は97%に向上し、この特定のコホートにおける最終的な4回の自殺企図すべてを成功裏に予測しました。
これらの挑発的な発見は現在、他のグループによって再現されており、エピジェネティックマーカーと自殺のような複雑で壊滅的なヒト行動を結びつける最も具体的な例の1つを提供しています。末梢バイオマーカーとヒトの精神病理および行動を関連付ける課題は、この研究で、末梢細胞における転写およびエピジェネティックプログラムが死後神経組織で見られるものに類似していること、および末梢におけるSKA2のメチル化率が朝のコルチゾールレベルおよび自殺念慮と関連していることを示すことによって見事に解決されました。最近、自殺念慮に対する迅速で有望な治療法として浮上しているケタミンによる治療によって、SKA2の末梢メチル化率が修飾され得るかどうかを検証することは興味深いでしょう。
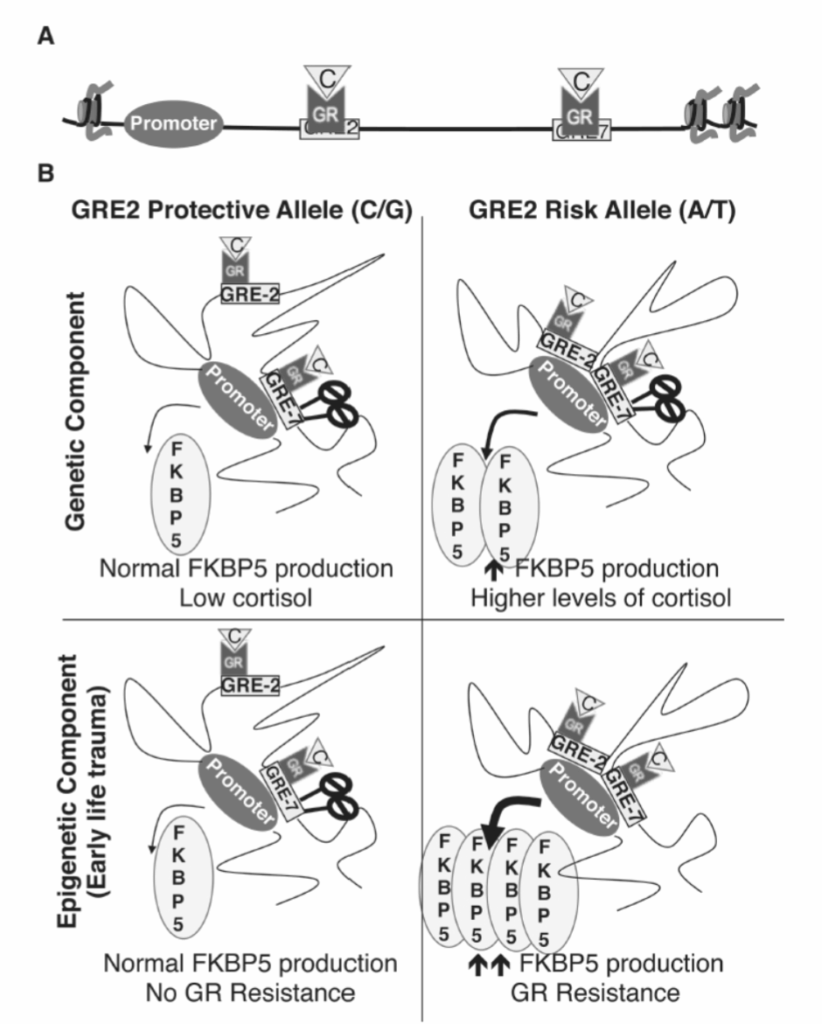
図 1.20-18. A: FKBP5遺伝子には、イントロン2(GRE-2)とイントロン7(GRE-7)に2つのGREが存在します。B: 上の2つのパネルは、FKBP5発現とGR抵抗性に対する遺伝的リスクの影響を示しています。左側では、保護的なC/Gアレルを含むGRE-2は、プロモーター/GRE-7複合体にリクルートされず、軽度のストレスに応答して比較的低いレベルのFKBP5と低いコルチゾールレベルを産生します。A/TアレルにGRE-2がプロモーター/GRE-7複合体に存在する場合(右上)、軽度のストレスに応答してより高いレベルのFKBP5と、結果としてより高いレベルのコルチゾールが産生されます。下のパネルは、FKBP5発現に対する幼少期ストレスのエピジェネティックな寄与を示しています。幼少期ストレスは、C/G保護アレルを持つ個人では、比較的低いレベルのコルチゾールとGRE-7の正常なメチル化をもたらします(左下パネル)。対照的に、幼少期ストレスは、A/Tリスクアレルを持つ個人のGRE-7のDNAメチル化を減少させ、より高いコルチゾールレベルにつながります。GRE-7の脱メチル化は、この部位へのGRのより効率的な結合をもたらし、したがって、より高いFKBP5レベルとGR抵抗性につながります(右下パネル)。
参考文献
- Cheung, S., Woo, J., Maes, M. S., & Zai, C. C. (2020). Suicide epigenetics, a review of recent progress. J Affect Disord, 265, 423-438.
- Ciabrelli, F., & Cavalli, G. (2015). Chromatin-driven behavior of topologically associating domains. J Mol Biol, 427, 608-625.
- Duffy, K. A., Bale, T. L., & Epperson, C. N. (2021). Germ cell drivers: transmission of preconception stress across generations. Front Hum Neurosci, 15, 642762.
- Guintivano, J., Brown, T., Newcomer, A., et al. (2014). Identification and replication of a combined epigenetic and genetic biomarker predicting suicide and suicidal behaviors. Am J Psychiatry, 171, 1287-1296.
- Guo, J. U., Ma, D. K., Mo, H., et al. (2011a). Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci, 14, 1345-1351.
- Guo, J. U., Su, Y., Zhong, C., Ming, G. L., & Song, H. (2011b). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell, 145, 423-434.
- Kaffman, A., & Meaney, M. J. (2007). Neurodevelopmental sequelae of postnatal maternal care in rodents: clinical and research implications of molecular insights. J Child Psychol Psychiatry, 48, 224-244.
- Kaminsky, Z. A., Tang, T., Wang, S. C., et al. (2009). DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet, 41, 240-245.
- Kim, Y., Lee, H. M., Xiong, Y., et al. (2017). Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader-Willi syndrome. Nat Med, 23, 213-222.
- Klengel, T., Mehta, D., Anacker, C., et al. (2013). Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci, 16, 33-41.
- Kouzarides, T. (2007). Chromatin modifications and their function. Cell, 128, 693-705.
- Kraan, C. M., Godler, D. E., & Amor, D. J. (2019). Epigenetics of fragile X syndrome and fragile X-related disorders. Dev Med Child Neurol, 61, 121-127.
- Kumar, A., Choi, K. H., Renthal, W., et al. (2005). Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron, 48, 303-314.
- Liu, X. S., Wu, H., Krzisch, M., et al. (2018). Rescue of fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell, 172, 979-992 e976.
- O’Donnell, K. J., & Meaney, M. J. (2020). Epigenetics, development, and psychopathology. Annu Rev Clin Psychol, 16, 327-350.
- Paris, J. (2006). Predicting and preventing suicide: do we know enough to do either? Harv Rev Psychiatry, 14, 233-240.
- Pena, C. J., Bagot, R. C., Labonte, B., & Nestler, E. J. (2014). Epigenetic signaling in psychiatric disorders. J Mol Biol, 426, 3389-3412.
- Peter, C. J., & Akbarian, S. (2011). Balancing histone methylation activities in psychiatric disorders. Trends Mol Med, 17, 372-379.
- Piatti, P., Zeilner, A., & Lusser, A. (2011). ATP-dependent chromatin remodeling factors and their roles in affecting nucleosome fiber composition. Int J Mol Sci, 12, 6544-6565.
- Reul, J. M., & Chandramohan, Y. (2007). Epigenetic mechanisms in stress-related memory formation. Psychoneuroendocrinology, 32 (Suppl 1), S21-25.
- Rogge, G. A., & Wood, M. A. (2013). The role of histone acetylation in cocaine-induced neural plasticity and behavior. Neuropsychopharmacology, 38, 94-110.
- Schafer, A. (2013). Gadd45 proteins: key players of repair-mediated DNA demethylation. Adv Exp Med Biol, 793, 35-50.
- Swygert, S. G., & Peterson, C. L. (2014). Chromatin dynamics: interplay between remodeling enzymes and histone modifications. Biochim Biophys Acta, 1839, 728-736.
- Tsankova, N. M., Berton, O., Renthal, W., Kumar, A., Neve, R. L., & Nestler, E. J. (2006). Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci, 9, 519-525.
- Vizan, P., Beringer, M., Ballare, C., & Di Croce, L. (2014). Role of PRC2-associated factors in stem cells and disease. FEBS J.
