
Saebom Lee, Jaehoon Jeong, Yongdo Kwak, Sang Ki Park*
Molecular Brain 2010: 3: 8
https://doi.org/10.1186/1756-6606-3-8
抄録
うつ病および関連する気分障害の分子基盤については、多様な仮説が提唱されてきたが、決定的な病因機序は未だに明確ではない。モノアミン仮説は、モノアミン系を標的とする抗うつ薬の有効性とともに、長きにわたってうつ病研究の中心的課題であった。抗うつ薬の効果の発現には急性のモノアミン系変化が関与していると広く考えられている一方で、現在の研究は、慢性的な抗うつ薬治療後に脳内で生じる長期的かつ下流の分子変化のメカニズムに焦点を移しつつある。このような背景のもと、本ミニレビューでは、うつ病および抗うつ薬の作用に関する分子レベルの視点に焦点を当て、気分障害の理解に向けた近年の主な研究テーマを簡潔に概説する。
序論
大うつ病や双極性障害といった気分障害は、現代社会において最も一般的な精神疾患である。人口の約16%が大うつ病を、約1%が双極性障害を生涯のうちに1回以上経験すると推定されている。これらに共通する症状群は「うつ病性症候群」と総称され、持続的な抑うつ気分、罪悪感、不安、死や自殺に関する反復思考などを含む。
うつ病の発症には、遺伝的要因が40~50%関与していると見積もられている。しかし、うつ病の多面的な症状を一つの遺伝子異常だけで引き起こすことは稀であり、複数の遺伝的因子が複合的に関与している可能性が高い。さらに、ストレス、情動的トラウマ、ウイルス感染、神経発達異常といった非遺伝的要因も、病態の複雑性を高めている。そのため、うつ病の分子機序に関する多様な仮説が提示されてきたが、確定的な病因メカニズムには未だ至っていない。
モノアミン仮説は、セロトニン、ドパミン、ノルアドレナリンといったモノアミン神経伝達物質の欠乏または不均衡がうつ病の原因であるとするものであり、過去50年にわたり研究の中心に位置してきた。この仮説は、三環系抗うつ薬やモノアミン酸化酵素阻害薬といった初期の抗うつ薬がモノアミン機能を急性に強化するという共通作用を持っていたことにより支持された。さらに、選択的セロトニン再取り込み阻害薬(SSRI)の開発により、仮説は一層強化された。
しかしながら、現在の抗うつ薬には未解決の問題が多い。第一に、抗うつ薬は全患者の50%未満にしか効果がなく、最近の新薬でも有効な患者の範囲を広げることには成功していない。第二に、臨床効果を得るには慢性的な投与が必要であり、その理由は不明である。第三に、抗うつ薬や気分安定薬は、多様な望ましくない副作用を有する。
特に、モノアミン系を急性に変化させる抗うつ薬の臨床効果が大幅に遅れて発現することから、慢性的な投与によって引き起こされる遺伝子発現などの持続的な下流変化が効果の本質であると考えられるようになってきた。この現象は、古典的なモノアミン仮説が想定したような単純な関係性では、うつ病を説明できないことを示唆している。うつ病は複数の病態を包含する疾患群であり、複数の神経伝達系の機能障害が相互に関係し合って症状を形成する可能性がある。
このように、抗うつ薬の効果発現にはモノアミン系の急性変化が介在することを踏まえつつも、現在の研究は慢性投与後の長期的かつ下流の分子メカニズムの解明へと焦点を移しており、うつ病および気分障害の病態生理の詳細な理解へと接近しつつある。本ミニレビューでは、うつ病と関連する気分障害の理解に向けた近年の主なアプローチをまとめる。
遺伝と環境の相互作用(Gene-environment interactions)
うつ病の素因となる遺伝子を見つける方法として、遺伝学者たちは長年にわたり、うつ病の発症における重要な環境要因であるライフストレスに対する反応に関与する遺伝子変異を探してきた。これは、**「遺伝と環境の相互作用」**の一例であり、環境因子が遺伝子の活動を介して作用することで、個人ごとにうつ病に対する感受性が異なるという考え方である。
この目的のために、セロトニントランスポーター(5-ヒドロキシトリプタミン輸送体、5-HTT)遺伝子の多型が広く研究されてきた。5-HTT遺伝子の**5’側フランキング領域(5-HTT遺伝子関連多型領域:5-HTTLPR)や、第2イントロンの可変数タンデムリピート(VNTR)**における多型が、5-HTTの発現レベルに影響を与えることが報告されている。
特に、5-HTTLPRの短鎖型(short variant)は、プロモーターの転写活性を抑制し、5-HTTの発現とセロトニン再取り込みを減少させることが知られており、長鎖型と比較される。
重要なことに、こうした多型がヒトの大うつ病性障害と関連することが遺伝学的研究で示されている。さらに、847名のニュージーランド人を対象にした縦断研究では、5-HTTLPRの短鎖アレルが、仕事の喪失や離婚などのライフストレスに応じたうつ病発症のリスク増加と関連していることが示された。
驚くべきことに、この研究では、有意なライフストレスが存在する場合にのみ、この多型の影響が顕在化することが分かり、5-HTTが遺伝的素因と環境的引き金との接点となりうる可能性が示唆された。
また、社会的支援を受けていない虐待歴のある子どもにおいて、5-HTTLPR短鎖型を有する群がうつ症状スコアの上昇を示すという研究もあり、これらの観察結果はさらに補強されている。
⚠️ 反証的知見と研究的限界
しかしながら、これらの研究結果から得られた洞察が他の研究で一貫して支持されているわけではない。
たとえば、5-HTT遺伝子のVNTR多型とうつ病感受性との関連は、一部の研究では検出されなかった。メタアナリシスでは、うつ病患者に5-HTTLPRと第2イントロンの多型が存在することは確認されたものの、その関連性の強さは統計的有意には達しなかった。
さらに、1206名の双生児を用いた大規模研究では、5-HTTLPRの主効果や、5-HTTLPRとストレスフルな出来事との交互作用は、うつ病との関連性が見られなかった。
最近の14件の研究を対象としたメタアナリシスでは、セロトニントランスポーターの遺伝型が単独、またはライフストレスとの相互作用として、うつ病のリスク上昇に関連するという証拠は得られなかったと結論づけられている。
これらの混在した研究結果は、特定の遺伝子変異に焦点を当てた「候補遺伝子アプローチ」の限界を浮き彫りにしており、特にうつ病のような多因子的疾患においては、偏りのない**全ゲノムスキャン(GWAS)**の重要性が改めて強調されている。
ストレス応答回路(Stress response circuits) 慢性的なストレスは、うつ病にとって重要な構成要素ではあるものの、それ自体が必要条件でも十分条件でもないように思われる。この観点から、視床下部-下垂体-副腎軸(HPA軸)、すなわち体内でストレスを調節する中枢内分泌回路は、うつ病研究における重要な関心対象となってきた【22】。
視床下部の傍室核(paraventricular nucleus)から分泌される副腎皮質刺激ホルモン放出因子(CRF)は、下垂体からの副腎皮質刺激ホルモン(ACTH)の分泌を促進する【22,23】。その後、副腎皮質からグルココルチコイドが分泌され、これは脳内のさまざまな領域における神経行動機能に影響を与える【2】。
このHPA軸は、海馬や扁桃体といった脳領域を介したフィードバックループを形成している【24】。報告によれば、**血中グルココルチコイドの持続的上昇(高コルチゾール血症)**は、海馬のCA3錐体ニューロンの興奮毒性を高め、樹状突起の萎縮、スパイン形成の減少、ニューロンのアポトーシス、さらには成人神経新生の阻害を引き起こす【25】。
このような慢性ストレスによって誘導される海馬ニューロンの機能異常は、HPA軸に対する抑制性の調節を減弱させ、結果としてHPA軸の過活動をもたらす【23】。
注目すべきは、うつ病患者の約半数でHPA軸の過活動が明らかとなっており、抗うつ薬による慢性治療によってこの過活動がしばしば逆転されるという点である【23,26】。さらに、動物研究では、慢性的な抗うつ薬投与によって神経新生が促進され、海馬の機能異常が回復することが示唆されている【27,28】。
治療戦略としてのHPA軸機能抑制の検討 このような知見に基づき、現在の研究の一方向として、HPA軸の機能を弱めることの治療的可能性を評価する試みが進んでいる。
明らかな標的としては、下垂体に発現するCRF受容体および、海馬その他の脳領域に発現するグルココルチコイド受容体が挙げられる。これらはHPA軸とそのフィードバックループの中核構成要素である【24,29–32】。
同様の文脈において、バソプレシン受容体も代替的標的として注目されている【33,34】。バソプレシンはCRFの機能を増強する神経ペプチドであり、扁桃体や大脳辺縁系の他部位に発現するバソプレシン受容体を介して作用する。
また、バソプレシン1b型(V1b)受容体の一塩基多型(SNP)が、大うつ病性障害に対する防御効果をもたらすことも報告されている【35】。
興味深いことに、CRF受容体・グルココルチコイド受容体・バソプレシン受容体に対する拮抗薬は、動物実験において抗うつ効果を示すようである。ただし、これがヒト患者への応用に耐えるかどうかは、今後の検討課題である。
神経栄養因子(Neurotrophic factors) 長期的なストレスは、**海馬における脳由来神経栄養因子(BDNF)の発現レベルを減少させるようである【36】。また、うつ病患者の剖検研究においても、BDNFの発現低下が報告されている【37】。さらに、BDNF遺伝子の多型は、うつ病の感受性を高める性格特性である神経症傾向(neuroticism)**と関連している【38】。
ある家族関連の関連研究では、BDNF遺伝子の多型が双極性障害と関連していることも示されている【39】。一方で、抗うつ薬の慢性投与は、BDNFレベルを高め、ストレス耐性を増強することが動物実験で示されている【40,41】。
これらの知見は、「神経栄養仮説(neurotrophism theory)」の基礎となっている。この仮説は、うつ病が神経栄養因子の欠乏によって引き起こされるとし、抗うつ薬はその欠乏を補うことで作用すると考えるものである。
この理論は、前述のストレス応答回路の過活動によって生じる海馬神経の損傷と密接に関係している可能性がある。BDNFは、脳内のさまざまな領域においてシナプス可塑性を促進することが知られているため、BDNF機能の改善は、ストレスによって傷つきやすい海馬神経にとって有益であると推察される【42,43】。
この考えを支持するように、BDNFを海馬へ直接注入した実験動物は、抗うつ薬投与と類似した行動変化を示すことが報告されている【41】。したがって、BDNFおよびその受容体であるTrkBは、新しいタイプの抗うつ療法の有望な標的として浮上してきている。
💡 困難と検討課題:因果関係の不明瞭さ これらの知見にもかかわらず、BDNF機能の変化がうつ病の病因や抗うつ薬の効果にどのように関与しているかについては、さらなる明確化が求められている。
たとえば、誘導性BDNFノックアウトマウスを用いた実験では、抗うつ薬の効果が抑制されたが、うつ病関連行動が観察されたのは雌のみであり、性差が顕著に見られた【36】。
さらに、前脳特異的TrkB受容体欠損マウスでは、強制水泳試験における行動的絶望感の増加といった、うつ病様行動が認められなかった【44】。しかし一方で、TrkB受容体の活性化は、抗うつ薬による行動効果に必要であることも示されている【45】。
したがって、BDNF活性の低下とうつ症状の出現は、単純な相関関係ではない可能性がある。とはいえ、神経栄養仮説が、新たな抗うつ療法の設計基盤として持つ潜在的価値は、現在の複雑な実験結果によって否定されるものではない。
ヒストン修飾(Histone modifications) 抗うつ薬のあまり理解されていない特徴の一つは、その効果が患者に現れるまでに長い遅延があるという点である【10】。この現象はしばしば、薬の有益な効果を支える神経の適応変化がゆっくり進行するためであると考えられている。
この「適応」の正体はまだ明らかになっていないが、**クロマチン状態の持続的な変化(=エピジェネティックな変化)**が関与していると考えられている。
たとえば、ある研究では、**一部のうつ病患者に有効である電気けいれん療法(electroconvulsive shocks)**が、実験動物において広範なヒストン修飾パターンの変化を引き起こすことが示された【46】。
特に顕著な変化が見られたのはBDNF遺伝子座であり、BDNFが新規抗うつ薬の標的として注目されていることと関連して、うつ症状の発現および慢性抗うつ薬治療におけるBDNFのエピジェネティック制御が精力的に研究されてきた。
ラットの海馬では、慢性的な電気けいれん療法によってBDNFプロモーター3および4におけるアセチル化ヒストンH3のレベルが上昇し、これがBDNFおよびCREBの発現増加と関連しているようである【46】。このアップレギュレーションは、抗うつ薬の効果とも関係づけられている【28,47】。
さらに、**慢性敗北ストレス(chronic defeat stress)**といううつ病モデルでは、海馬内の特定のBDNFスプライスバリアントの発現が選択的に低下することが報告されている【28】。
この発現抑制は、**ヒストンH3-K27の二重メチル化(転写抑制のヒストンコード)**の誘導によると考えられている【28,48】。
一方で、抗うつ薬治療によってBDNF発現の抑制が逆転されることも示されており、これは**ヒストンH3のアセチル化およびH3-K4のメチル化(いずれも転写活性化を示すコード)**の誘導による可能性が高い【49】。
🔧 ヒストン脱アセチル化酵素(HDAC)の役割 この一連の過程において、ヒストン脱アセチル化酵素(HDAC)の役割は非常に重要であると考えられている。
具体的には、慢性抗うつ薬投与はHDAC5をダウンレギュレートし、HDAC5を海馬に過剰発現させると抗うつ薬の効果が阻害されることが報告されている【28】。
そのため、HDAC阻害薬は、うつ病および関連する気分障害に対する有望な新規治療薬として注目されている。
HDAC阻害薬は、以下の4つの化学的ファミリーに分類される:
短鎖脂肪酸(例:酪酸ナトリウム(SB)、フェニル酪酸、バルプロ酸(VPA))
ヒドロキシアミン酸(例:TSA、サブエロイルアニリドヒドロキサム酸(SAHA))
エポキシケトン類(例:トラポキシン)
ベンズアミド類
なかでも、最も広く使用されている気分安定薬の一つが**バルプロ酸(VPA)**である。
VPAは、HDAC1および他のHDACに対する阻害活性を持つことが知られており【50】、その気分安定化作用がヒストン修飾を介している可能性が提案されている。
さらに、VPA、SB、TSAといったHDAC阻害薬が、脳内のBDNF発現を増加させるという研究もある【51】。
このように、エピジェネティック機構、特にヒストン修飾は、うつ病の病態理解と新規治療法開発に向けた新しい機序的洞察を提供しうる重要な領域である。
成人海馬神経新生(Adult hippocampal neurogenesis) うつ病患者における海馬体積の減少を示す脳画像研究は、成人期の神経新生がうつ病の病態と関係している可能性を探るきっかけとなった【52】。この仮説によれば、慢性的ストレスやその他のうつ病誘発因子は神経新生を減少させる一方で、抗うつ薬の有効性は神経新生の増加に依存している可能性があるとされる【53–56】。
成人の神経新生は、**側脳室下帯(subventricular zone)および海馬歯状回の顆粒層下帯(subgranular zone)**に限局しており【57】、このことは、うつ病の発症および治療の過程における海馬神経新生の重要性を強く示唆している。
この考えを支持するように、うつ病モデル動物(学習性無力感、慢性軽度ストレス、心理社会的ストレスなど)では、海馬神経新生の減少が一貫して観察されている【58–60】。一方で、抗うつ薬の慢性投与は、神経新生を増加させるだけでなく、新生ニューロンの生存を支持することが示されている【61】。
また、三環系抗うつ薬であるイミプラミンや、**選択的セロトニン再取り込み阻害薬(SSRIs)**の効果は、齧歯類の海馬神経新生を必要とすることも報告されている【58,62,63】。
さらに、慢性フルオキセチン投与は、錐体細胞層のシナプス数を増加させ、歯状回およびその他の海馬細胞層でのスパイン密度の減少を防ぐことが示唆されている【64】。
注目すべきは、海馬神経新生を促進することが知られている“豊かな環境(enriched environments)”が、齧歯類におけるうつ様行動を軽減するという研究結果である【65–67】。
🔬 BDNFと神経新生の分子メカニズム 神経新生の増加を仲介する抗うつ薬の分子メカニズムを探る際には、BDNFの発現レベルに注目すべきである。
上述のように、様々なうつ病モデル動物では、BDNFレベルが低下している【40】。一方で、慢性的な抗うつ薬投与や電気けいれん療法は、海馬におけるBDNFレベルを上昇させる【28,46】。
最近の研究では、BDNFを含むCRE配列をもつ標的遺伝子の発現を調節する転写因子であるCREBが、抗うつ薬の慢性投与によって海馬でアップレギュレートおよび活性化されることが示されている【2,53,68,69】。
しかしながら、CREBやBDNFの誘導、神経新生、抗うつ薬による行動効果の因果関係は、今後さらなる検討を要する。
💊 気分安定薬と神経新生 最近の研究では、リチウム、バルプロ酸、カルバマゼピンといった気分安定薬の長期投与が、成人海馬神経新生を増加させることも示されている【70–72】。
リチウムは、グリコーゲン合成酵素キナーゼ-3(GSK-3)およびイノシトールシグナル伝達を直接阻害する【73】。
**バルプロ酸(VPA)**は、HDACの阻害によって遺伝子発現を増強し、間接的にGSK-3活性を阻害し、イノシトールシグナル伝達を抑制する【71,74–76】。
これらの経路が、気分安定薬の臨床効果と実際に関連しているかどうかはまだ不明であるが、データは、成人海馬神経新生へと収束する共通の分子経路が、うつ病と関連気分障害の病態生理の中核にある可能性を示唆している。
薬物離脱(Substance withdrawal) アルコール、ニコチン、ベンゾジアゼピン、コカイン、オピオイドなどの物質の乱用と依存は、うつ病の高リスクと関連している。特に、薬物の中断や離脱に続いて発症するうつ症状は、よく知られており、依存症治療の大きな課題である【77】。
動物モデルにおいても、これらの薬物の慢性投与後の急激な中止は、「うつ様」行動(depression-like behavior)を誘導することが示されている【78,79】。これは例えば、強制水泳試験や味覚嫌悪条件づけ、社会的相互作用テストなどで観察される。
薬物離脱に伴ううつ様行動は、行動的無力感、報酬感受性の低下、社会的回避、食欲低下などの形で現れ、臨床で報告されるうつ症状と類似している。
🧠 神経生物学的基盤 この種の「うつ様」行動の基盤には、報酬系回路の機能不全が関与していると考えられている。たとえば、**中脳辺縁ドパミン系(ventral tegmental area から側坐核への経路)**は、快感と報酬処理の中枢であり、薬物投与によって一時的に活性化されるが、離脱後には著しく活動が抑制される【80】。
この活動の低下は、報酬関連行動の減少と負の情動反応の増加を引き起こし、うつ様行動に寄与する。
さらに、**慢性的な薬物使用後には、神経可塑性関連遺伝子(例:BDNF、CREB、ΔFosB)**の発現にも変化が見られ、脳内の報酬回路の恒常性維持機構が破綻することが示唆されている【81】。
💊 治療への含意 興味深いことに、抗うつ薬が薬物離脱後のうつ様行動を軽減することが、いくつかの動物実験で示されている【82】。
たとえば、慢性フルオキセチン投与は、モルヒネ離脱後のうつ様行動を有意に軽減し、
三環系抗うつ薬であるデシプラミンは、アルコール離脱時のうつ様行動とストレス応答を緩和する。
これらの結果は、薬物離脱に伴ううつ様状態が、少なくとも部分的には古典的なうつ病と共通の分子経路に依存している可能性を示している。
結論と今後の展望(Conclusions and future directions)
うつ病およびその他の気分障害に関する研究は、過去数十年間で著しく進展した。とりわけ、分子神経生物学的視点からの探究により、これらの疾患の病因や治療機序に関する有望な手がかりが得られている。
かつては、モノアミン仮説が中心的な理論であったが、近年では、神経栄養因子、ストレス応答回路、遺伝と環境の相互作用、エピジェネティクス、成人神経新生といった多層的な生物学的機構が注目されるようになった。
これらの知見に基づいて、現在では抗うつ薬の作用は単にモノアミン濃度の上昇によるものではなく、慢性的な投与により誘導される神経可塑性の変化や遺伝子発現の修飾に依存していると理解されている。
また、うつ病の病因が単一の神経伝達物質や脳領域の異常だけでは説明できないという認識が広まりつつある。うつ病は、複雑で多因子的な疾患群であり、その背景には遺伝的素因、環境的ストレス因子、個体の発達歴、神経免疫応答、そしてエピジェネティックな制御が交錯していると考えられている。
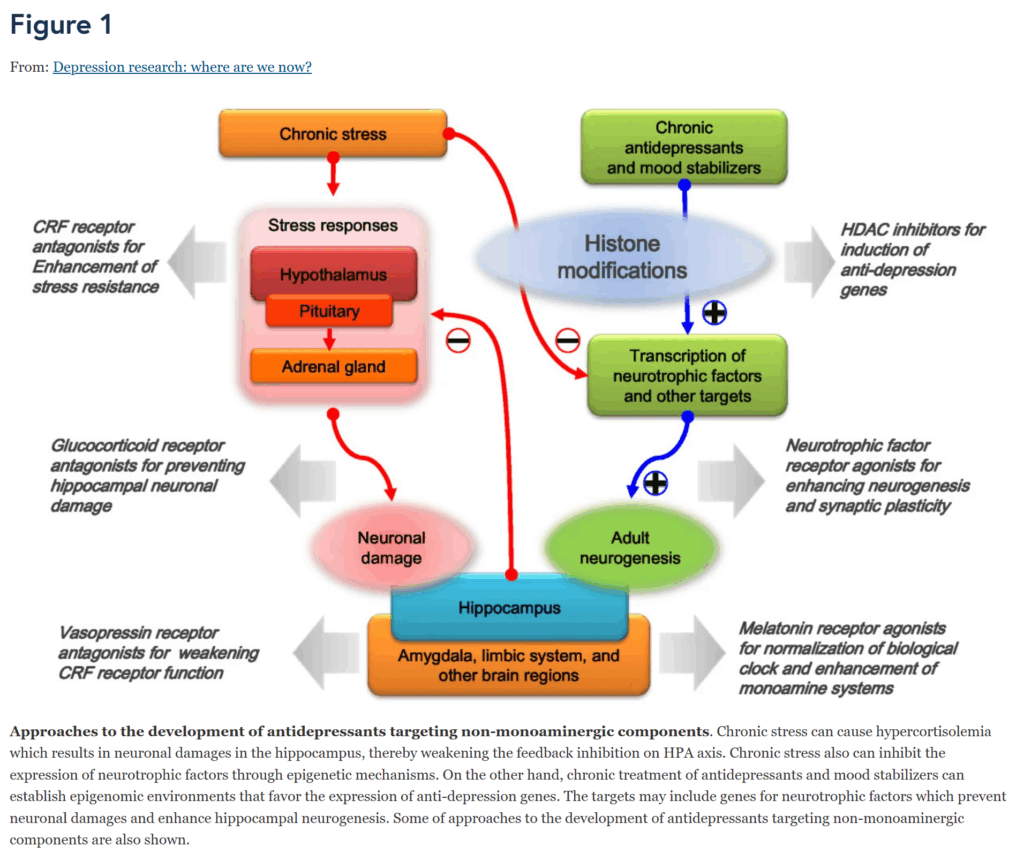
図1 非モノアミン性成分を標的とする抗うつ薬開発のアプローチ
慢性ストレスは高コルチゾール血症を引き起こし、それが海馬の神経細胞損傷をもたらし、それによってHPA軸へのフィードバック阻害を弱めます。慢性ストレスはまた、エピジェネティックなメカニズムを通じて神経栄養因子の発現を抑制することもあります。一方で、抗うつ薬や気分安定薬の慢性的な治療は、抗うつ遺伝子の発現を促進するエピゲノム環境を確立することができます。その標的には、神経細胞の損傷を防ぎ、海馬の神経新生を促進する神経栄養因子の遺伝子が含まれる可能性があります。非モノアミン性成分を標的とする抗うつ薬開発のアプローチの一部も示されています。
🔭 今後の研究の方向性
今後の研究課題としては、以下のようなテーマが挙げられる:
- より精緻な疾患モデルの開発
― 単純な行動指標では捉えきれない「人間の抑うつ状態」に迫る動物モデルや、疾患サブタイプに対応した系統的な実験系の整備が求められる。 - 大規模ゲノム解析とエピゲノム解析の統合
― ゲノムワイド関連解析(GWAS)やエピゲノム編集技術を用いて、うつ病の分子基盤に関するより包括的な理解を目指すことができる。 - 個別化医療とバイオマーカーの確立
― BDNFレベルや神経新生マーカー、エピジェネティック指標などを用いて、治療反応性や予後を予測する精密医療の基盤が構築されつつある。 - 新規治療標的の探索
― HDAC阻害薬、GSK-3阻害薬、グルタミン酸系修飾薬、ニューロステロイドなど、モノアミン系に依存しない抗うつ治療薬の開発が進んでいる。 - 環境介入・行動療法との併用研究
― 環境の豊かさや社会的相互作用が脳に与える影響を統合的に捉えることで、心理社会的介入と生物学的治療の統合戦略が開発される可能性がある。
うつ病の理解は、今まさに還元主義からネットワーク的理解へと転換しつつある。神経科学、遺伝学、心理学、社会学の融合的研究が、うつ病という複雑な人間の苦悩に対して、より深く、より本質的に迫る鍵となるだろう。
参考文献
- Krishnan V, Nestler EJ (2008) The molecular neurobiology of depression. Nature 455(7215):894–902. doi:10.1038/nature07455
- Duman RS, Monteggia LM (2006) A neurotrophic model for stress-related mood disorders. Biol Psychiatry 59(12):1116–1127.
- Hyman SE (2007) Can neuroscience be integrated into the DSM-V? Nat Rev Neurosci 8(9):725–732.
- Berton O, Nestler EJ (2006) New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci 7(2):137–151.
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002) Neurobiology of depression. Neuron 34(1):13–25.
- Manji HK, Quiroz JA, Sporn J, Payne JL, Denicoff K, Gray NA, Zarate CA, Charney DS (2003) Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry 53(8):707–742.
- Belmaker RH, Agam G (2008) Major depressive disorder. N Engl J Med 358(1):55–68.
- Charney DS, Manji HK (2004) Life stress, genes, and depression: multiple pathways lead to increased risk and new opportunities for intervention. Sci STKE 2004(225):re5.
- Krishnan V, Nestler EJ (2008) Linking molecules to mood: new insight into the biology of depression. Am J Psychiatry 165(10):1202–1214.
- Duman RS (2004) Role of neurotrophic factors in the etiology and treatment of mood disorders. Neuromolecular Med 5(1):11–25.
- Sullivan PF, Neale MC, Kendler KS (2000) Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 157(10):1552–1562.
- Kendler KS, Gardner CO, Prescott CA (2002) Toward a comprehensive developmental model for major depression in women. Am J Psychiatry 159(7):1133–1145.
- Hirschfeld RM (1999) The Comorbidity of Major Depression and Anxiety Disorders: Recognition and Management in Primary Care. Prim Care Companion J Clin Psychiatry 1(6):244–254.
- Hasler G, Drevets WC, Manji HK, Charney DS (2004) Discovering endophenotypes for major depression. Neuropsychopharmacology 29(10):1765–1781.
- Nestler EJ, Carlezon WA Jr (2006) The mesolimbic dopamine reward circuit in depression. Biol Psychiatry 59(12):1151–1159.
- Hariri AR, Holmes A (2006) Genetics of emotional regulation: the role of the serotonin transporter in neural function. Trends Cogn Sci 10(4):182–191.
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R (2003) Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301(5631):386–389.
- Kaufman J, Yang BZ, Douglas-Palumberi H, Houshyar S, Lipschitz D, Krystal JH, Gelernter J (2004) Social supports and serotonin transporter gene moderate depression in maltreated children. Proc Natl Acad Sci USA 101(49):17316–17321.
- Gillespie NA, Whitfield JB, Williams B, Heath AC, Martin NG (2005) The relationship between stressful life events, the serotonin transporter (5-HTTLPR) genotype and major depression. Psychol Med 35(1):101–111.
- Munafo MR, Clark TG, Roberts KH, Johnstone EC (2006) Neuroticism mediates the association of the serotonin transporter gene with lifetime major depression. Neuropsychobiology 53(1):1–8.
- Risch N, Herrell R, Lehner T, Liang KY, Eaves L, Hoh J, Griem A, Kovacs M, Ott J, Merikangas KR (2009) Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA 301(23):2462–2471.
- de Kloet, E. R., Joels, M., & Holsboer, F. (2005). Stress and the brain: from adaptation to disease. Nature Reviews Neuroscience, 6(6), 463–475.
- Parker, K. J., Schatzberg, A. F., & Lyons, D. M. (2003). Neuroendocrine aspects of hypercortisolism in major depression. Hormones and Behavior, 43(1), 60–66.
- Brown, E. S., Varghese, F. P., & McEwen, B. S. (2004). Association of depression with medical illness: does cortisol play a role? Biological Psychiatry, 55(1), 1–9.
- McEwen, B. S. (2007). Physiology and neurobiology of stress and adaptation: central role of the brain. Physiological Reviews, 87(3), 873–904.
- Raison, C. L., & Miller, A. H. (2003). When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. American Journal of Psychiatry, 160(9), 1554–1565.
- Scaccianoce, S., Del Bianco, P., Paolone, G., Caprioli, D., Modafferi, A. M., Nencini, P., & Badiani, A. (2006). Social isolation selectively reduces hippocampal brain-derived neurotrophic factor without altering plasma corticosterone. Behavioural Brain Research, 168(2), 323–325.
- Tsankova, N. M., Berton, O., Renthal, W., Kumar, A., Neve, R. L., & Nestler, E. J. (2006). Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nature Neuroscience, 9(4), 519–525.
- Holsboer, F. (2000). The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology, 23(5), 477–501.
- Valdez, G. R. (2009). CRF receptors as a potential target in the development of novel pharmacotherapies for depression. Current Pharmaceutical Design, 15(14), 1587–1594.
- Todorovic, C., Sherrin, T., Pitts, M., Hippel, C., Rayner, M., & Spiess, J. (2009). Suppression of the MEK/ERK signaling pathway reverses depression-like behaviors of CRF2-deficient mice. Neuropsychopharmacology, 34(5), 1416–1426.
- Skelton, K. H., Oren, D., Gutman, D. A., Easterling, K., Holtzman, S. G., Nemeroff, C. B., & Owens, M. J. (2008). The CRF1 receptor antagonist, R121919, attenuates the severity of precipitated morphine withdrawal. European Journal of Pharmacology, 571(1), 17–24.
- Simon, N. G., Guillon, C., Fabio, K., Heindel, N. D., Lu, S. F., Miller, M., … & Koppel, G. A. (2008). Vasopressin antagonists as anxiolytics and antidepressants: recent developments. Recent Patents on CNS Drug Discovery, 3(2), 77–93.
- Surget, A., & Belzung, C. (2008). Involvement of vasopressin in affective disorders. European Journal of Pharmacology, 583(2-3), 340–349.
- van West, D., Del-Favero, J., Aulchenko, Y., Oswald, P., Souery, D., Forsgren, T., … & Claes, S. J. (2004). A major SNP haplotype of the arginine vasopressin 1B receptor protects against recurrent major depression. Molecular Psychiatry, 9(3), 287–292.
- Monteggia, L. M., Luikart, B., Barrot, M., Theobold, D., Malkovska, I., Nef, S., … & Nestler, E. J. (2007). Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biological Psychiatry, 61(2), 187–197.
- Karege, F., Vaudan, G., Schwald, M., Perroud, N., & La Harpe, R. (2005). Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Research. Molecular Brain Research, 136(1–2), 29–37.
- Sen, S., Nesse, R. M., Stoltenberg, S. F., Li, S., Gleiberman, L., Chakravarti, A., … & Burmeister, M. (2003). A BDNF coding variant is associated with the NEO personality inventory domain neuroticism, a risk factor for depression. Neuropsychopharmacology, 28(2), 397–401.
- Neves-Pereira, M., Mundo, E., Muglia, P., King, N., Macciardi, F., & Kennedy, J. L. (2002). The brain-derived neurotrophic factor gene confers susceptibility to bipolar disorder: evidence from a family-based association study. American Journal of Human Genetics, 71(3), 651–655.
- Duman, R. S., & Monteggia, L. M. (2006). A neurotrophic model for stress-related mood disorders. Biological Psychiatry, 59(12), 1116–1127.
- Shirayama, Y., Chen, A. C., Nakagawa, S., Russell, D. S., & Duman, R. S. (2002). Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. Journal of Neuroscience, 22(8), 3251–3261.
- Sairanen, M., Lucas, G., Ernfors, P., Castrén, M., & Castrén, E. (2005). Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. Journal of Neuroscience, 25(5), 1089–1094.
- Berton, O., McClung, C. A., Dileone, R. J., Krishnan, V., Renthal, W., Russo, S. J., … & Nestler, E. J. (2006). Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science, 311(5762), 864–868.
- Zörner, B., Wolfer, D. P., Brandis, D., Kretz, O., Zacher, C., Madani, R., … & Gass, P. (2003). Forebrain-specific trkB-receptor knockout mice: behaviorally more hyperactive than “depressive”. Biological Psychiatry, 54(9), 972–982.
- Saarelainen, T., Hendolin, P., Lucas, G., Koponen, E., Sairanen, M., MacDonald, E., … & Castrén, E. (2003). Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. Journal of Neuroscience, 23(1), 349–357.
- Tsankova, N. M., Kumar, A., & Nestler, E. J. (2004). Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. Journal of Neuroscience, 24(24), 5603–5610.
- Wilkinson, M. B., Xiao, G., Kumar, A., LaPlant, Q., Renthal, W., Sikder, D., … & Nestler, E. J. (2009). Imipramine treatment and resiliency exhibit similar chromatin regulation in the mouse nucleus accumbens in depression models. Journal of Neuroscience, 29(24), 7820–7832.
- Jenuwein, T., & Allis, C. D. (2001). Translating the histone code. Science, 293(5532), 1074–1080.
- Tsankova, N., Renthal, W., Kumar, A., & Nestler, E. J. (2007). Epigenetic regulation in psychiatric disorders. Nature Reviews Neuroscience, 8(5), 355–367.
- Yasuda, S., Liang, M. H., Marinova, Z., Yahyavi, A., & Chuang, D. M. (2009). The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Molecular Psychiatry, 14(1), 51–59.
- Schroeder, F. A., Lin, C. L., Crusio, W. E., & Akbarian, S. (2007). Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biological Psychiatry, 62(1), 55–64.
- Drevets, W. C. (2001). Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Current Opinion in Neurobiology, 11(2), 240–249.
- Pittenger, C., & Duman, R. S. (2008). Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology, 33(1), 88–109.
- Sahay, A., & Hen, R. (2007). Adult hippocampal neurogenesis in depression. Nature Neuroscience, 10(9), 1110–1115.
- Dranovsky, A., & Hen, R. (2006). Hippocampal neurogenesis: regulation by stress and antidepressants. Biological Psychiatry, 59(12), 1136–1143.
- Hunsberger, J. G., Newton, S. S., Bennett, A. H., Duman, C. H., Russell, D. S., Salton, S. R., & Duman, R. S. (2007). Antidepressant actions of the exercise-regulated gene VGF. Nature Medicine, 13(12), 1476–1482.
- Gage, F. H. (2002). Neurogenesis in the adult brain. Journal of Neuroscience, 22(3), 612–613.
- Surget, A., Saxe, M., Leman, S., Ibarguen-Vargas, Y., Chalon, S., Griebel, G., Hen, R., & Belzung, C. (2008). Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biological Psychiatry, 64(4), 293–301.
- Lee, K. J., Kim, S. J., Kim, S. W., Choi, S. H., Shin, Y. C., Park, S. H., Moon, B. H., Cho, E., Lee, M. S., Chun, B. G., & Shin, K. H. (2006). Chronic mild stress decreases survival, but not proliferation, of new-born cells in adult rat hippocampus. Experimental & Molecular Medicine, 38(1), 44–54.
- Vollmayr, B., Simonis, C., Weber, S., Gass, P., & Henn, F. (2003). Reduced cell proliferation in the dentate gyrus is not correlated with the development of learned helplessness. Biological Psychiatry, 54(9), 1035–1040.
- Santarelli, L., Saxe, M., Gross, C., Surget, A., Battaglia, F., Dulawa, S., Weisstaub, N., Lee, J., Duman, R., Arancio, O., Belzung, C., & Hen, R. (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science, 301(5634), 805–809.
- Chen, F., Madsen, T. M., Wegener, G., & Nyengaard, J. R. (2009). Imipramine treatment increases the number of hippocampal synapses and neurons in a genetic animal model of depression. Hippocampus, 20(12), 1376–1384.
- Boldrini, M., Underwood, M. D., Hen, R., Rosoklija, G. B., Dwork, A. J., Mann, J. J., & Arango, V. (2009). Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology, 34(11), 2376–2389.
- Wu, X., & Castrén, E. (2009). Co-treatment with diazepam prevents the effects of fluoxetine on the proliferation and survival of hippocampal dentate granule cells. Biological Psychiatry, 66(1), 5–8.
- Kempermann, G., Kuhn, H. G., & Gage, F. H. (1997). More hippocampal neurons in adult mice living in an enriched environment. Nature, 386(6624), 493–495.
- Brenes Saenz, J. C., Villagra, O. R., & Fornaguera Trías, J. (2006). Factor analysis of Forced Swimming test, Sucrose Preference test and Open Field test on enriched, social and isolated reared rats. Behavioural Brain Research, 169(1), 57–65.
- Hattori, S., Hashimoto, R., Miyakawa, T., Yamanaka, H., Maeno, H., Wada, K., & Kunugi, H. (2007). Enriched environments influence depression-related behavior in adult mice and the survival of newborn cells in their hippocampi. Behavioural Brain Research, 180(1), 69–76.
- Boer, U., Alejel, T., Beimesche, S., Cierny, I., Krause, D., Knepel, W., & Flügge, G. (2007). CRE/CREB-driven up-regulation of gene expression by chronic social stress in CRE-luciferase transgenic mice: reversal by antidepressant treatment. PLoS ONE, 2(5), e431.
- Blendy, J. A. (2006). The role of CREB in depression and antidepressant treatment. Biological Psychiatry, 59(12), 1144–1150.
- Chen, G., Zeng, W. Z., Yuan, P. X., Huang, L. D., Jiang, Y. M., Zhao, Z. H., & Manji, H. K. (1999). The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. Journal of Neurochemistry, 72(2), 879–882.
- Leng, Y., Liang, M. H., Ren, M., Marinova, Z., Leeds, P., & Chuang, D. M. (2008). Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase-3 inhibition. Journal of Neuroscience, 28(10), 2576–2588.
- Kalemenev, S. V., Kosacheva, E. S., Sem’ianov, A. V., Godukhin, O. V., & Raevskii, K. S. (1998). Effect of anticonvulsants lamotrigine and carbamazepine on the synaptic transmission in CA1 field of the rat hippocampal slices. Biulleten’ Eksperimental’noi Biologii i Meditsiny, 126(9), 307–310.
- Jope, R. S. (2003). Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends in Pharmacological Sciences, 24(9), 441–443.
- Kim, A. J., Shi, Y., Austin, R. C., & Werstuck, G. H. (2005). Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. Journal of Cell Science, 118(1), 89–99.
- Kim, H. J., Rowe, M., Ren, M., Hong, J. S., Chen, P. S., & Chuang, D. M. (2007). Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. Journal of Pharmacology and Experimental Therapeutics, 321(3), 892–901.
- Chen, G., Huang, L. D., Jiang, Y. M., & Manji, H. K. (1999). The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. Journal of Neurochemistry, 72(3), 1327–1330.
- Koob, G. F. (2000). Animal models of craving for ethanol. Addiction, 95(Suppl 2), S73–S81.
- Barr, A. M., Markou, A., & Phillips, A. G. (2002). A ‘crash’ course on psychostimulant withdrawal as a model of depression. Trends in Pharmacological Sciences, 23(10), 475–482.
- Taylor, J. R., Punch, L. J., & Elsworth, J. D. (1998). A comparison of the effects of clonidine and CNQX infusion into the locus coeruleus and the amygdala on naloxone-precipitated opiate withdrawal in the rat. Psychopharmacology, 138(2), 133–142.
- Spielewoy, C., & Markou, A. (2003). Withdrawal from chronic phencyclidine treatment induces long-lasting depression in brain reward function. Neuropsychopharmacology, 28(6), 1106–1116.
- Kampman, K. M., Volpicelli, J. R., McGinnis, D. E., Alterman, A. I., Weinrieb, R. M., D’Angelo, L., & Epperson, L. E. (1998). Reliability and validity of the Cocaine Selective Severity Assessment. Addictive Behaviors, 23(4), 449–461.
- Gawin, F. H., & Kleber, H. D. (1986). Abstinence symptomatology and psychiatric diagnosis in cocaine abusers. Clinical observations. Archives of General Psychiatry, 43(2), 107–113.
- Uslaner, J., Kalechstein, A., Richter, T., Ling, W., & Newton, T. (1999). Association of depressive symptoms during abstinence with the subjective high produced by cocaine. American Journal of Psychiatry, 156(9), 1444–1446.
- Plotsky, P. M., Owens, M. J., & Nemeroff, C. B. (1998). Psychoneuroendocrinology of depression. Hypothalamic-pituitary-adrenal axis. Psychiatric Clinics of North America, 21(2), 293–307.
- Deuschle, M., Schweiger, U., Weber, B., Gotthardt, U., Korner, A., Schmider, J., … & Heuser, I. (1997). Diurnal activity and pulsatility of the hypothalamus-pituitary-adrenal system in male depressed patients and healthy controls. Journal of Clinical Endocrinology & Metabolism, 82(1), 234–238.
- London, E. D., Simon, S. L., Berman, S. M., Mandelkern, M. A., Lichtman, A. M., Bramen, J., … & Ling, W. (2004). Mood disturbances and regional cerebral metabolic abnormalities in recently abstinent methamphetamine abusers. Archives of General Psychiatry, 61(1), 73–84.
- Nestler, E. J., & Carlezon, W. A. Jr. (2006). The mesolimbic dopamine reward circuit in depression. Biological Psychiatry, 59(12), 1151–1159.
- Scully, J. H. (2000). The American Psychiatric Association textbook of psychiatry (3rd ed.). Journal of Clinical Psychiatry, 61(4), 306.
- Park, S. K., Nguyen, M. D., Fischer, A., Luke, M. P., Affar, el B., Dieffenbach, P. B., … & Tsai, L. H. (2005). Par-4 links dopamine signaling and depression. Cell, 122(2), 275–287.
- Tanda, K., Nishi, A., Matsuo, N., Nakanishi, K., Yamasaki, N., Sugimoto, T., … & Miyakawa, T. (2009). Abnormal social behavior, hyperactivity, impaired remote spatial memory, and increased D1-mediated dopaminergic signaling in neuronal nitric oxide synthase knockout mice. Molecular Brain, 2, 19.
- Mark, G. P., Kinney, A. E., Grubb, M. C., & Keys, A. S. (1999). Involvement of acetylcholine in the nucleus accumbens in cocaine reinforcement. Annals of the New York Academy of Sciences, 877, 792–795.
- Verhoeff, N. P., Christensen, B. K., Hussey, D., Lee, M., Papatheodorou, G., Kopala, L., … & Kapur, S. (2003). Effects of catecholamine depletion on D2 receptor binding, mood, and attentiveness in humans: a replication study. Pharmacology Biochemistry and Behavior, 74(2), 425–432.
- Solomon, R. L., & Corbit, J. D. (1974). An opponent-process theory of motivation. I. Temporal dynamics of affect. Psychological Review, 81(2), 119–145.
- American Psychiatric Association. (2000). Diagnostic and statistical manual of mental disorders: DSM-IV-TR (4th ed.). Washington, DC: American Psychiatric Association.
- Quello, S. B., Brady, K. T., & Sonne, S. C. (2005). Mood disorders and substance use disorder: a complex comorbidity. Science & Practice Perspectives, 3(1), 13–21.
- Laasonen-Balk, T., Kuikka, J., Viinamäki, H., Husso-Saastamoinen, M., Lehtonen, J., & Tiihonen, J. (1999). Striatal dopamine transporter density in major depression. Psychopharmacology, 144(3), 282–285.
- Jacobsen, L. K., Staley, J. K., Malison, R. T., Zoghbi, S. S., Seibyl, J. P., Kosten, T. R., & Innis, R. B. (2000). Elevated central serotonin transporter binding availability in acutely abstinent cocaine-dependent patients. American Journal of Psychiatry, 157(7), 1134–1140.
- Malison, R. T., Price, L. H., Berman, R., van Dyck, C. H., Pelton, G. H., Carpenter, L., … & Charney, D. S. (1998). Reduced brain serotonin transporter availability in major depression as measured by [123I]β-CIT SPECT. Biological Psychiatry, 44(11), 1090–1098.
- Nunes, E. V., McGrath, P. J., Quitkin, F. M., & Wager, S. (2006). Antidepressant treatment of depression in patients with substance use disorders. In H. R. Kranzler & B. S. Rounsaville (Eds.), Dual diagnosis and treatment: Substance abuse and comorbid medical and psychiatric disorders (pp. 231–270). New York: Marcel Dekker.
- Lee, S., Jeong, J., Kwak, Y., & Park, S. K. (2010). Depression research: where are we now? Molecular Brain, 3, 8. https://doi.org/10.1186/1756-6606-3-8
